
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Role of the environment in the stability of anisotropic gold particles
Robinson
Cortes-Huerto†
*abc,
Jacek
Goniakowski
ab and
Claudine
Noguera
ab
aCNRS, UMR 7588, Institut des Nanosciences de Paris, F-75005 Paris, France
bSorbonne Universités, UPMC University Paris 06, UMR 7588, INSP, F-75005 Paris, France
cCEA, UMR 3299, LIONS, DSM, IRAMIS, SIS2M, F-91191 Gif Sur Yvette, France. E-mail: corteshu@mpip-mainz.mpg.de
First published on 27th January 2015
Abstract
Despite the long-lasting interest in the synthesis control of nanoparticles (NPs) in both fundamental and applied nanosciences, the driving mechanisms responsible for their size and shape selectivity in an environment (solution) are not completely understood, and a clear assessment of the respective roles of equilibrium thermodynamics and growth kinetics is still missing. In this study, relying on an efficient atomistic computational approach, we decipher the dependence of energetics, shapes and morphologies of gold NPs on the strength and nature of the metal–environment interaction. We highlight the conditions under which the energy difference between isotropic and elongated gold NPs is reduced, thus prompting their thermodynamic coexistence. The study encompasses both monocrystalline and multi-twinned particles and extends over size ranges particularly representative of the nucleation and early growth stages. Computational results are further rationalized with arguments involving the dependence of facet and edge energies on the metal–environment interactions. We argue that by determining the abundance and diversity of particles nucleated in solution, thermodynamics may constitute an important bias influencing their final shape. The present results provide firm grounds for kinetic simulations of particle growth.
1 Introduction
One of the great challenges in nanosciences is to produce size- and shape-selected nanoparticles (NPs) in a controlled way.1 This stems from the strong dependence of their properties on these structural characteristics. Among others is the enhanced rate of catalytic reactions as the specific surface area of a NP increases, or the variability of optical properties as a function of electron confinement and/or NP morphology. For example, ZnO NPs have proved to be able to adopt a very wide range of morphologies, such as nanocombs, nanorings, nanohelices, nanosprings, nanobelts, nanowires, nanocages, bipods, tetrapods, etc.,2 to be exploited in new generation devices. Similarly, gold or silver NPs have received much attention in the last few years due to their potential applications in chemical sensing, biological imaging, drug delivery and phototherapeutics.3–5 As a consequence, efforts have been made to synthesize them with controlled morphologies, such as spheres,6,7 nanorods,8–13 pentagonally twinned particles, nanoplatelets,14 or ultra-thin wires.15Shape-selected NPs may be obtained by a variety of techniques, ranging from wet chemical methods to physical vapor deposition, metal–organic chemical vapor deposition (MOCVD), molecular beam epitaxy (MBE), etc.16 The size and shape selection may depend on various factors related to the gaseous or liquid environment they are in contact with, among which are temperature, gas pressure or solution supersaturation, and the presence of capping/surfactant agents. It is far from being clear whether thermodynamics or kinetics drive the final shape and what is the mechanism responsible for symmetry breaking,17 when the latter takes place. From a theoretical point of view, kinetics is often invoked but rarely quantified. Thermodynamics has been explored in very specific cases. For example, adsorption of noble gas,18 CO,19–21 or thiolate22 onto gold NPs has been shown to modify the surface energies and thus the Wulff shapes, most often leading to an increase of sphericity of the particles. Adsorption of halides, silver and surfactants onto gold nanorods, on the other hand, was suggested to promote particle elongation.23–25
More generally, the identification of equilibrium shapes and structures of NPs in a given environment is a prerequisite to assess the origin of experimentally observed anisotropic NPs and provide firm grounds for any valid analysis of the role of kinetics- and growth-driven effects. Indeed, at thermodynamic equilibrium, the abundance of a given type of particles scales exponentially with their excess energy, according to the Boltzmann distribution. As a consequence, the range of particle types increases rapidly when the dispersion of their excess energies diminishes. The same is true in the nucleation stage where the nucleation barrier is proportional to the excess energy and where the anisotropies of critical nuclei may give rise to a large variety of growth shapes. It is important to stress that standard tools used to determine NP shapes, such as the Wulff construction, may become insufficient and/or inadequate when the surface/edge energy ratio is strongly altered by interaction with the environment and/or when multi-twinned particles are formed.
In this context, the present atomistic study focuses on the thermodynamic stability of different NP families, representative of both monocrystalline (MC) and multi-twinned (MT) structures, with both isotropic and anisotropic (elongated or flattened) shapes. Aside from being a preliminary step to future full-scale dynamic simulations of particle growth, its goal is to highlight mechanisms responsible for a reduction of the NP energy dispersion as a function of the nature and strength of the metal–environment interaction, and to rationalize the trends in terms of the competition between edge and facet energies. Moreover, analyzing the evolution of the particle stability as a function of size, new thermodynamic effects in the small size regime are evidenced, relevant to the nucleation stage. Far beyond the arguments of Wulff, such a study is made possible, thanks to the efficient energetic approach for the metal–environment interaction,26 allowing the structure and shape optimization of NPs in the size ranging from a few tens up to several thousands of atoms, while mimicking interaction types ranging from covalent-like, through pair-wise, up to strongly coordination-dependent.
The paper is organized in the following way. After a description of the Model and methods in Section 2, a diagram of surface stability is given as a function of the parameters controlling the gold–environment interaction, and the relevant NP facet orientations are identified (Section 3). Then, relying on atomistic simulations, the effect of the environment on the stability of large particles is highlighted (Section 4) and size effects are described (Section 5). A rationale for the computed trends is discussed in terms of facet and edge energies (Section 6) and conclusions are given (Section 7).
2 Model and methods
Due to the large number of atoms typically involved in NPs in contact with gaseous or liquid environments, a light energetic model is required to allow an efficient simulation of such systems. Following our previous work,26 we describe metal–metal interactions via a many-body expression derived from the second-moment approximation (SMA) to the tight-binding model27–31 and the metal–environment interaction by an additional many-body potential which modifies the energy of the outermost NP atoms in a way which depends on their coordination. In doing so, the species of the environment are not explicitly treated, which alleviates the simulation cost.More precisely, the total energy E(N) of the system of N metallic atoms is written as a sum over all atoms i in the following way:
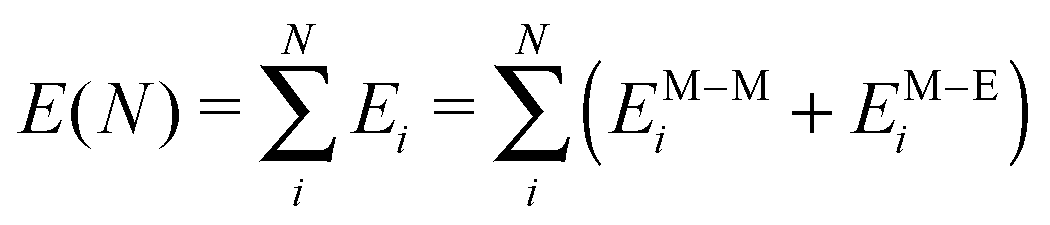 | (1) |
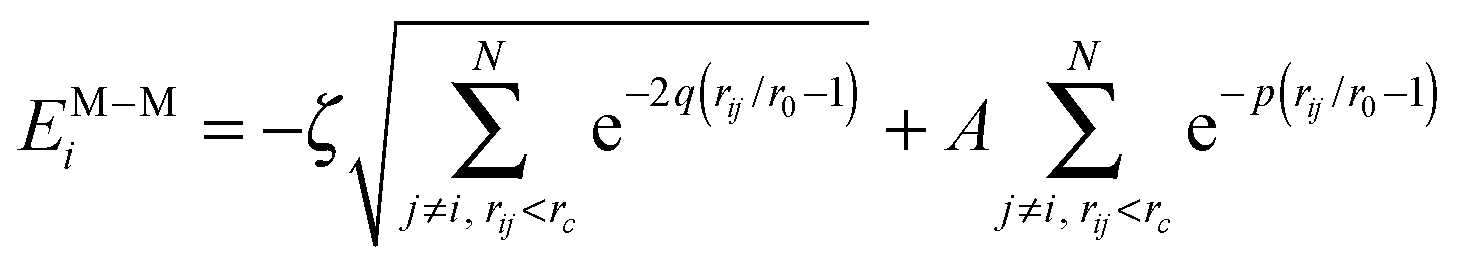 | (2) |
The interaction between a surface atom of coordination Zi (ZB = 12 is the bulk coordination in gold) and the environment, EM−Ei in eqn (1), is represented by a coordination-dependent energy term:26
| EM−Ei = −εnpi | (3) |
In the present implementation of our model, particularly suited to the quenched molecular dynamics used in motif optimization of particle shape and morphology (see below), Zi does not change during the dynamics and thus it has been simply defined as the number of first Au–Au neighbors (the Au–Au distance smaller than midway between the first and second neighbors in the bulk Au lattice). Additionally, we introduce Ze as the maximum number of bonds that an isolated metal atom can form with the environmental atoms/molecules. For example, in solution, Ze represents the solvation number. Typically, Ze ≤ 8 for water depending upon the size and charge of the metallic atom, whereas Ze ≪ 8 for solvents with larger molecules. Thus eqn (3) applies as such for atoms of coordination Zi ≥ ZB − Ze. For less coordinated Au atoms, EM−Ei = −ε(ZB − Ze)p. In the present study on gold NPs we systematically assume Ze = 7, which produces different strengths of interaction with the environment for gold atoms with Zi ≥ 5.
With this energetic model, the motif optimization approach35 is used to generate families of isotropic, elongated or flattened closed-shell NPs. First, a variety of initial NPs are generated by a sequence of different rigid truncations of monocrystalline (MC) and multi-twinned (MT) structures, involving different proportions of selected facets, and a single high-symmetry elongation axis, which is {001} for MC particles and the five-fold axis for MT particles. Each of these NPs is then brought to its local energy minimum by quenched molecular dynamics (time step 7 fs), with the initial temperature T ∼ 300 K, tuned so as to preserve the NP core structure, as well as the type and proportion of the exposed facets. The resulting NPs are then assembled into distinct families, according to their length-to-diameter aspect ratio R = 2Iz/(Ix + Iy), estimated from the components of the diagonalized tensor of inertia I.36 The most stable particles in each family are then identified by their lowest excess energy:35
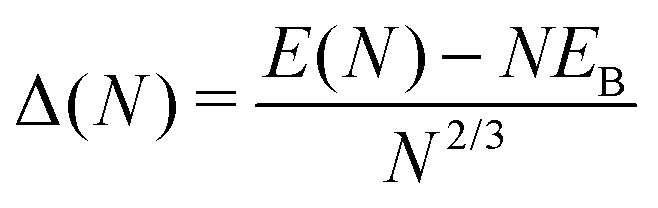 | (4) |
3 Surface energies of gold in contact with the environment
As a prerequisite for the understanding of NP characteristics, this section focuses on the relative stability of semi-infinite gold surfaces in various environments. Fig. 1 displays a map of the most stable surface orientations as a function of (ε, p), obtained from calculations performed on about 40 inequivalent surfaces,26 including both low-index surfaces and a variety of high-index surfaces, with coordination numbers of surface atoms ranging from Z = 9, as at the (111) surface, down to Z = 5 at multi-atomic step edges at (nn0) surfaces (n > 1). As expected, negative surface energies are found in the top right part of the diagram (white region in Fig. 1) and the dense (111) surface is found to be the most stable in its bottom left part, ranging from vacuum (ε = 0) up to the limit at which the surface energy becomes negative. Only in environments strongly sensitive to the coordination number (p > 1), the (100) (Zi = 8) and, to a lesser extent, (210) surface orientations (with its 6-, 9- and 11-fold coordinated atoms) are favored.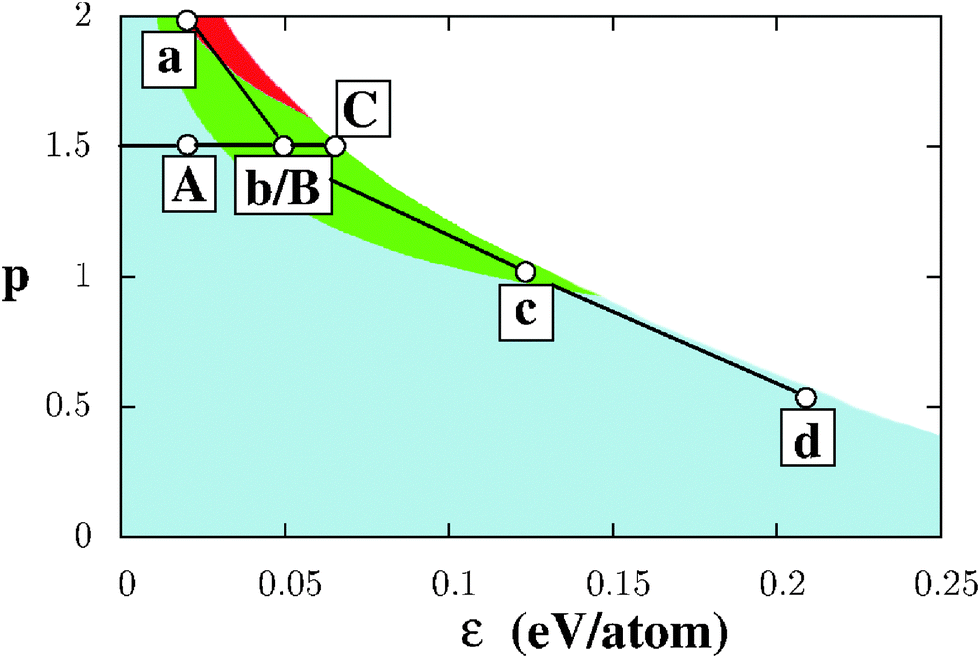 | ||
| Fig. 1 Color map showing the most stable surface orientations as a function of environment conditions (ε, p). Surface energies are negative in the white region. The light-gray (blue), gray (green), and dark-gray (red) colors represent regions where (111), (100), and (210) surfaces, respectively, are the most stable. The two series of points, (A)–(C) and (a)–(d), represent selected environments used in the present study. Interconnecting lines are guides to the eye. | ||
In the considered (ε, p) range we have selected two series of environments, Fig. 1. Series I consists of environments Vac (vacuum), (A), (B), and (C), associated with a fixed value of p (p = 1.5) and variable ε values, homogeneously spanning the range from ε = 0 (Vac) up to the largest value compatible with the positive surface energies ε = 0.06 eV per atom (C). This series enables an explicit analysis of the effect of the interaction strength ε and of the change in the most stable facet orientation from (111) to (100) on the NP morphologies. Conversely, series II consists of conditions (a), (b), (c), and (d) for which the nature of the metal–environment bonding varies from interactions strongly sensitive to atom coordination in (a) and (b) (p ≥ 1.5), through the first-neighbor pair-wise interactions (p = 1) in (c), to covalent-line interactions (p = 1/2) in (d). These four environments have been purposely chosen close to the limit of negative surface energies, so as to make the impact of interaction with the environment better pronounced. Series I and II, which share a single common environment, denoted hereafter (b/B), will be systematically used throughout the paper to study the effect of the environment on the NP stability and shape.
Beyond the information on the most stable surfaces, Fig. 2 displays the surface energies σhkl for all considered orientations as a function of the total number of broken bonds per unit area, for the two selected series of environments. Coherently with the stabilizing environmental effect, all surface energies are shifted downwards on the energy scale with respect to vacuum conditions. This is particularly true for environment (C) and for all environments of the second series. While, in vacuum, surface energies increase as a function of the number of broken bonds; this increase is progressively attenuated [(A)], then flattened [(b/B) and (C)]. Moreover, the dispersion of σhkl for surfaces with a similar number of broken bonds per surface area increases as ε increases, driven by the non-linear character of EM−Ei. These two effects produce numerous environment-driven stability inversions between surfaces of different orientations and, in particular, make the (100) surface the most stable in (b/B) and (C) environments and the (210) one in (a), consistent with Fig. 1.
 | ||
| Fig. 2 Energies of low- and various high-index surfaces as a function of the number of broken bonds per unit surface area, calculated for model environment conditions indicated in Fig. 1. Top panel: Series I [environments: Vac, (A), (b/B), and (C)]; bottom panel: Series II [environments: (a), (b/B), (c), and (d)]. | ||
Aside from the three low-index surfaces (111), (100) and (110), the (210) surface appears among the lowest energy terminations. Since the energy differences between various surfaces in the family of (n10) vicinal orientations (n > 1) are close to the limit of precision of the present model, we will consider the (210) surface as a representative of this whole family. In an analogous way, the (311) surface (Z = 7 and 10) will be considered as representative of the vicinal (n11) family. In the following study we will systematically use the set of (111), (110), (100), (210), and (311) truncations in the structural motif optimization approach.
4 Results: large nanoparticles
This section presents the results issued from motif optimization of particles which are relatively “large” (size N ∼ 3000 atoms), albeit in the nanoscale range. It successively focuses on the variations of the strength (ε parameter) of metal–environment interactions (Section 4.1), the nature (p parameter) of the metal–environment bonding (Section 4.2), and finally provides results regarding the relative stability of MC versus MT particles (Section 4.3).4.1 Strength of the metal–environment interaction
Fig. 3 and 4 display the variation of excess energy differences in Series I [Vac, (A), (b/B), and (C) in Fig. 1], as a function of the strength ε of the metal–environment interaction, between the most stable isotropic (R = 1) and anisotropic (R ≠ 1) MC and MT particles, respectively. The corresponding atomistic representations are shown in the lower panels of these figures.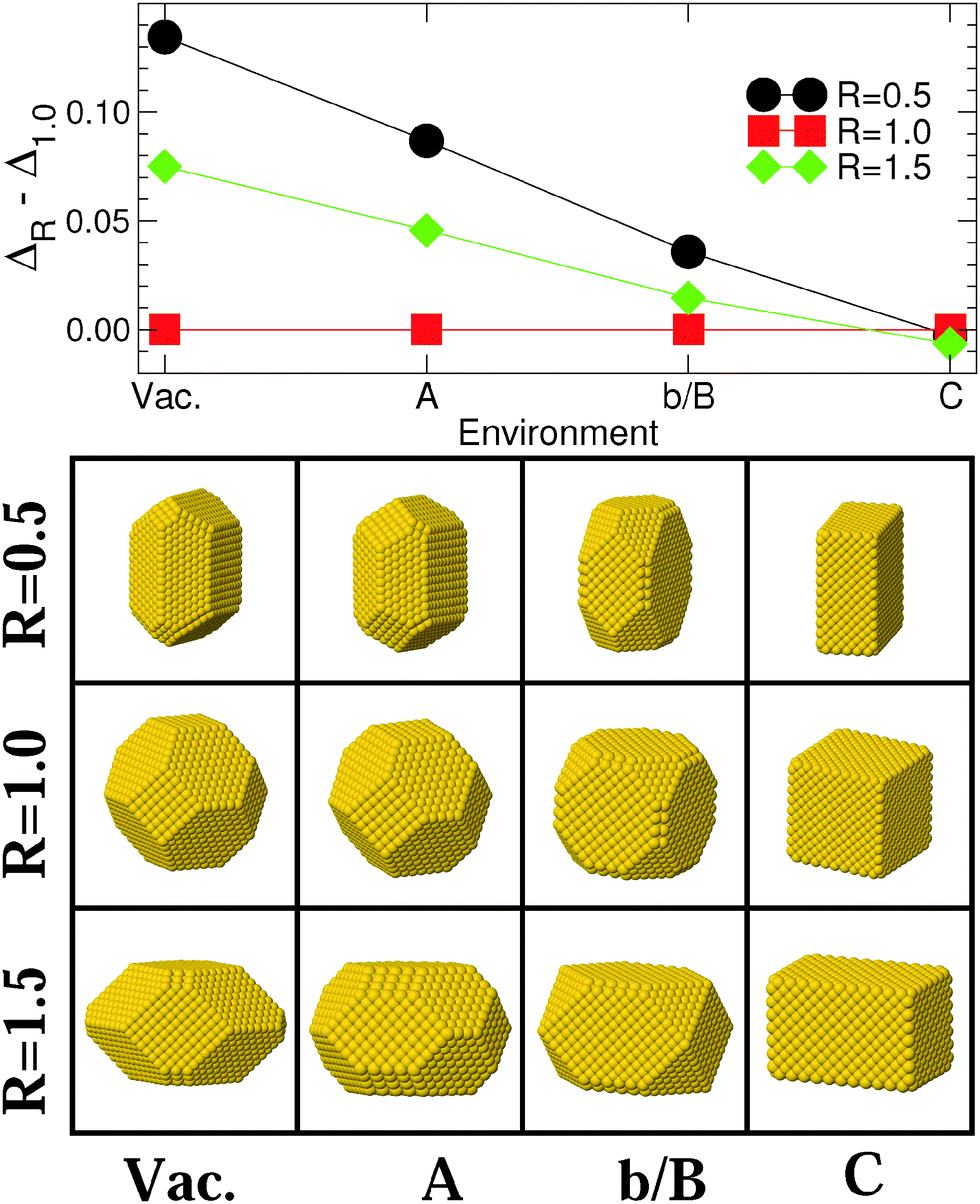 | ||
| Fig. 3 Upper panel: differences in excess energies ΔR − Δ1.0 (eV/N2/3) between the most stable MC particles (N ≈ 3000), with aspect ratios R = 0.5, 1.5 and 1.0, in the four environments of Series I [Vac, (A), (b/B), and (C) of Fig. 1]. Lines are guides to the eye. Lower panel: atomic representation of the corresponding NPs. | ||
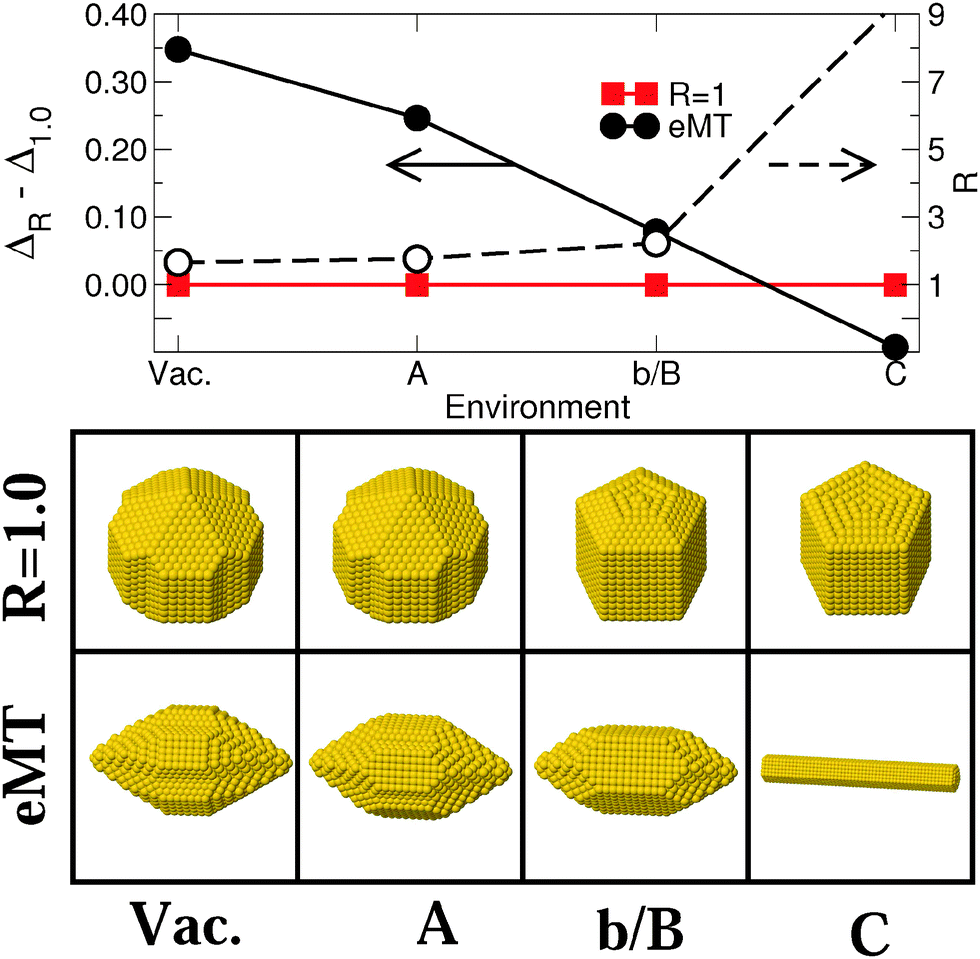 | ||
| Fig. 4 Upper panel: differences in excess energies ΔR − Δ1.0 (eV/N2/3) between isotropic (R = 1, full squares) and elongated (R > 1, full circles) most stable elongated MT particles (eMT) (N ≈ 3000) and the aspect ratio of the latter (empty circles) in the four environments of Series I [Vac, (A), (b/B), and (C) in Fig. 1]. Lines are guides to the eye. Left and right arrows refer to the data for energy and aspect ratio scales, respectively. Lower panel: atomic representation of the corresponding NPs. | ||
The morphologies of isotropic (R = 1), elongated (R = 1.5) and flattened (R = 0.5) MC NPs follow similar trends (Fig. 3), with a progressive change from (111)-terminated shapes with only small (100) truncations (Vac) to (100)-terminated cuboids with smaller and smaller (111) facets as ε increases. While, under vacuum conditions, isotropic MC NPs are by far the most stable; the excess energy difference ΔR − Δ1.0 diminishes progressively as ε increases, due principally to the overall decrease of surface and edge energies (Fig. 2). Eventually, a stability inversion occurs in (C) where both types of anisotropic NPs (R = 0.5 and 1.5) are energetically favored.
Fig. 4 reports the corresponding results obtained for two families of MT particles, one with an aspect ratio equal to 1 and the other consisting of particles elongated along the five-fold axis (R > 1.0). For the latter, the value of R which minimizes Δ has been determined for each environment. In vacuum, isotropic MT NPs expose five large (111) facets at each extremity and small (111) and (100) lateral facets. The size of the terminal (111) facets decreases as ε increases, while the lateral (100) truncations grow larger. The same is true for the lateral (100) facets of elongated particles, while the facets at their tips evolve from (311) and (110) in (Vac) to (110) only in (A), (b/B) and (C). As for MC particles, the energy dispersion between isotropic and elongated MT (eMT) particles diminishes progressively with an increase in ε. ΔR − Δ1.0 changes sign in environment (C).
To summarize, increasing the strength ε of the metal–environment interaction induces a change in the shape and energetics of the NPs. The orientation of the prevailing facets moves from (111) to (100) in MC NPs and the tip facets of MTs evolve from (111) to (311) and (110). Moreover, the increase of ε induces a strong overall decrease of excess energy dispersion between particles of different shape and aspect ratio. In the extreme case of the most strongly interacting environment (C), this leads to a stability inversion between isotropic and anisotropic NPs.
4.2 Nature of the metal–environment interaction
Fig. 5 and 6 report results regarding the stability and shape of MC and MT particles, respectively, along Series II of environments [(a), (b/B), (c), and (d) in Fig. 1], which differ by the nature of the metal–environment interaction from strongly coordination-dependent [environments (a) and (b/B)], through pair-wise [(c)], to covalent-like [(d)]. Both MC and MT particles evolve in a qualitatively similar way along this series, (210) and (100) facets are dominant in environment (a), the importance of (210) decreases in (b/B) to the benefit of (111) facets, although the latter remain small. Finally, the proportion of (111) facets increases in environment (c) and even more in (d). This sequence closely follows the trends expected from the diagram (ε, p) of the most stable surfaces, Fig. 1.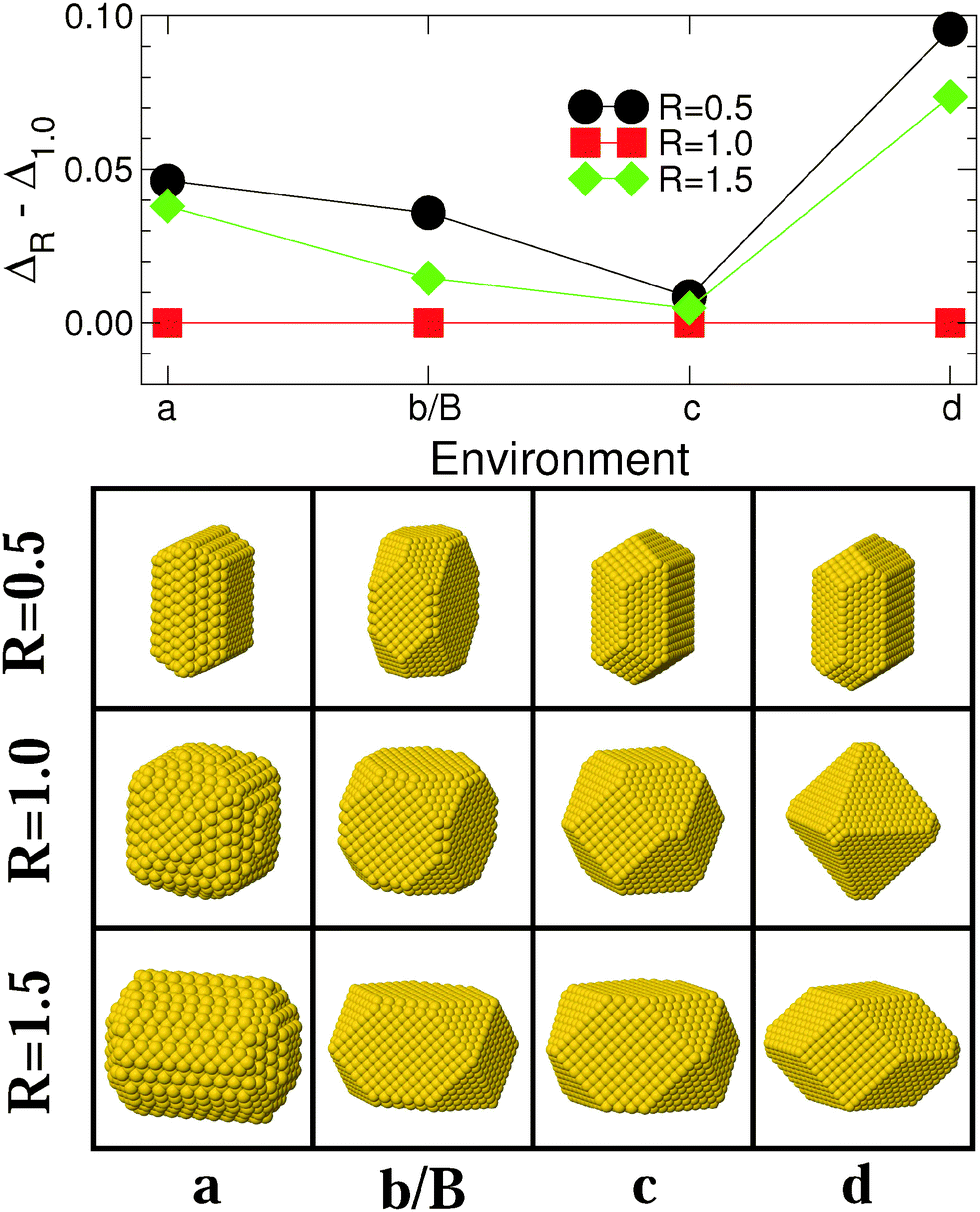 | ||
| Fig. 5 Same as Fig. 3 for MC particles interacting with environments of Series II [(a), (b/B), (c), and (d) in Fig. 1]. Lines are guides to the eye. | ||
 | ||
| Fig. 6 Same as Fig. 4 for MT particles interacting with environments of Series II [(a), (b/B), (c), and (d) in Fig. 1]. Lines are guides to the eye. Left and right arrows refer to the data for energy and aspect ratio scales, respectively. | ||
From the energetic point of view, for MC particles, we find a progressive reduction of the excess energy dispersion when going from (a) to (c). Conversely, environment (d) is by far the least favorable to anisotropic shapes. A similar strong preference for isotropic (R = 1) particles in environment (d) is found for MT NPs, Fig. 6. For these MT particles, the excess energy difference is especially small in the very particular case of environment (a), likely due to the fact that a small elongation (R ≈ 2) allows them to be terminated by the large, most stable (210) facets.
To summarize, the nature of the metal–environment interaction impacts the excess energy dispersion between isotropic and anisotropic NPs. For MC particles, a pair-wise metal–environment interaction [p ∼ 1.0, environment (c)] provides the optimal conditions for an efficient reduction of the energy gap between particles of different shapes and for the stabilization of anisotropic NPs. Conversely, the relative stability of elongated MT particles is optimal in the most strongly-coordination dependent environment (a). For both types of particles, elongated shapes are expected to be the least abundant when the metal–environment interaction is covalent-like (d).
4.3 Relative stability of MC and MT nanoparticles
Fig. 7 compares the excess energies of MC and MT particles in Series I and II. Since in most cases, isotropic MC NPs are the most stable, they are used as a common reference in each of the environments.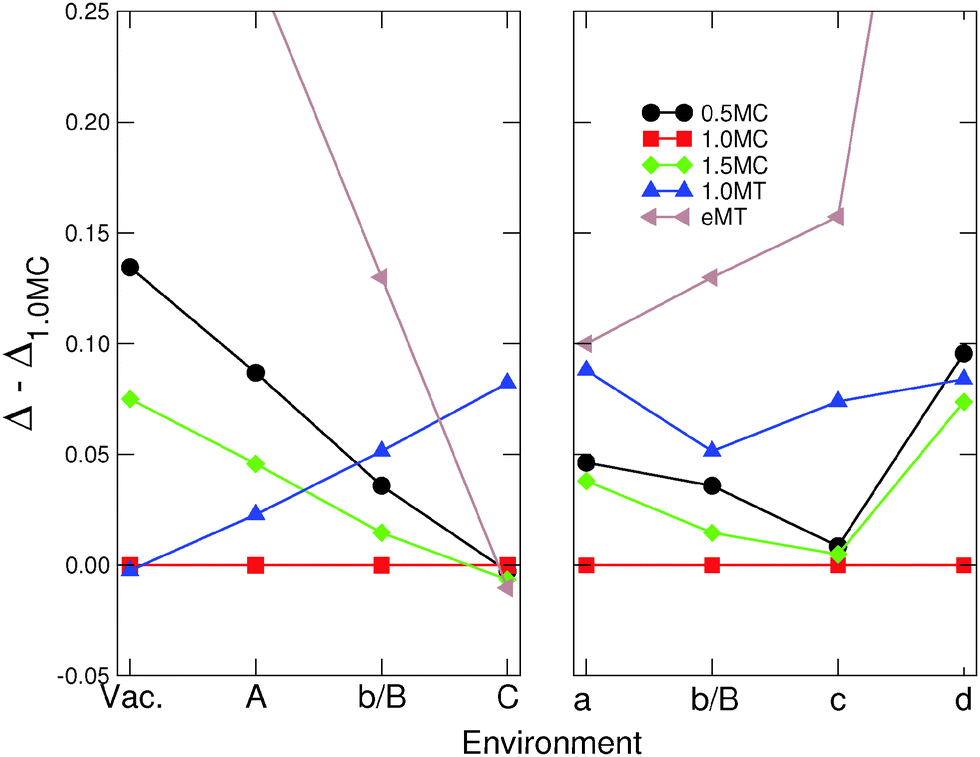 | ||
| Fig. 7 Differences in excess energies ΔR − Δ1.0 between different families of MC and MT particles in the environments of Series I (left panel) and Series II (right panel). The excess energy of the most stable isotropic MC particle (R = 1.0) is used as a reference in each environment. Lines are guides to the eye. | ||
Isotropic MT particles are found to be energetically favored over the MC ones only under vacuum conditions, where a high proportion of (111) facets for a minimal particle volume is the main reason of their stability at relatively small sizes.37 With a growing metal–environment interaction strength (Series I) the isotropic MT particles become progressively destabilized. Conversely, the excess energy of the elongated MT particles, decreases rapidly along Series I. It is worth noticing that this decrease is much more rapid than that of anisotropic MC particles, which results in a net thermodynamic preference for elongated MT particles in an extreme environment (C).
In Series II the reduction of excess energy dispersion between MT and MC particles is not as pronounced. We find that strongly coordination-dependent environments [(a) and (b/B)] produce a somewhat smaller dispersion, but the effect is relatively weak. Conversely, the covalent-like environment (d) is visibly less efficient for the stabilization of elongated MT.
To summarize, aside from vacuum conditions, MT NPs are, in most environments, less stable than their (isotropic) MC counterparts. We find however that strongly coordination-dependent environments efficiently stabilize the elongated MT NPs, which may in extreme cases become thermodynamically favored over isotropic and anisotropic MC particles.
5 Size effects
Beyond the results regarding “large” NPs presented above, this section focuses on other environment-induced effects, explicitly driven by their “small” size (smaller than 1500 atoms). For simplicity we will restrict the presentation only to the three families of closed-atomic-shell MC particles (R = 0.5, 1.0, and 1.5) in the scalable regime, and to environments (a), (b/B), and (c) of Series II.Fig. 8 reports the dependence of excess energies on NP sizes, in the range N = 50–1500 atoms. As a reference the corresponding data for N ∼ 3000 are recalled, as well as the results obtained for MC particles in vacuum. In the four depicted cases, the relative stability order and dispersion of the largest (N ∼ 1500) and most stable particles of the three families correspond closely to those obtained for the particles of size N ∼ 3000. This shows that the main size-driven effects indeed take place for N < 1500 atoms.
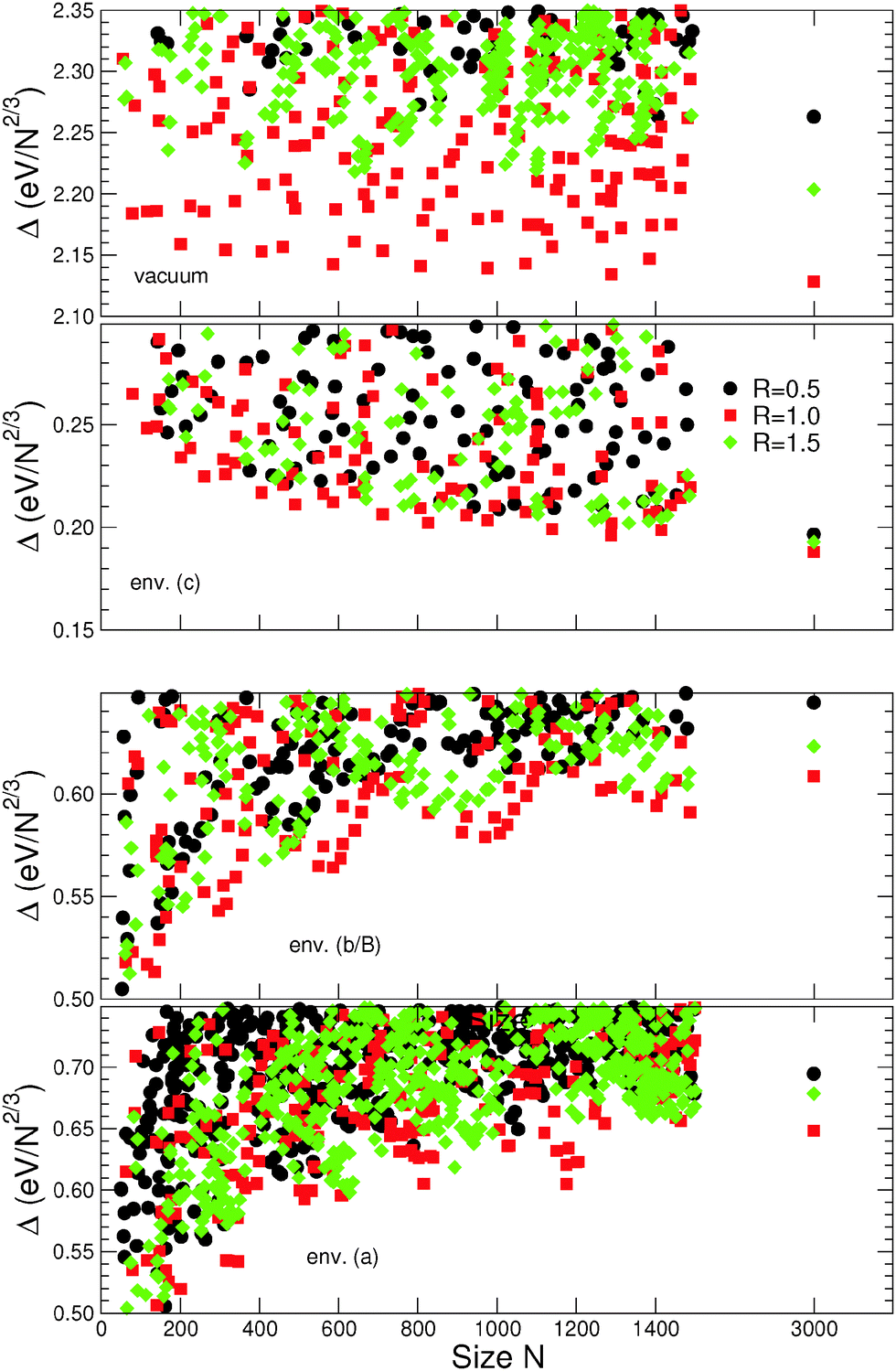 | ||
| Fig. 8 Excess energy Δ(N) (eV/N2/3) as a function of size (50 < N < 1500 atoms) for MC particles with aspect ratios R = 0.5 (black symbols), 1.0 (red symbols), and 1.5 (green symbols) in vacuum and in environments (c), (b/B), and (a) of Series II. The results obtained for large particles (N ∼ 3000 atoms) are recalled for the sake of reference in the right part of each panel. | ||
Superimposed on an overall monotonic variation with size, the excess energies Δ(N) for the most stable NPs of each family display an oscillatory behavior, characterized by local minima at discrete values of N, well pronounced in environments (a) and (b/B) and systematically deeper for smaller particles.
In vacuum, since the isotropic NPs are by far the most stable, the oscillatory character of Δ(N) does not impact substantially the energy dispersion between different types of particles. In environment (c), the behavior of Δ(N) in each family is regular and its oscillation amplitude is smaller than the excess energy difference between the three families. The situation is qualitatively different in environments (a) and (b/B), where the oscillations of Δ(N) as a function of size become well pronounced. In these cases, the energy dispersion between the three families is strongly size-dependent, as exemplified by the series of stability inversions between isotropic (R = 1.0) and elongated (R = 1.5) particles. For example, in environment (b/B), a well pronounced thermodynamic preference for elongated particles appears at sizes N ∼ 200 and 400 atoms, while at N ∼ 800 and 1300 atoms isotropic and elongated particles are quasi-degenerate.
To summarize, the existence of a strong, size-driven modulation of the excess energy dispersion between isotropic and anisotropic particles has been highlighted. It concerns principally smaller particles (typically, N < 1000 atoms) in environments characterized by a strong dependence on the coordination of metal atoms (p > 1). In such environments there exist size ranges in which elongated particles are quasi-degenerate with or even more stability than the isotropic ones.
6 Discussion
The results obtained for the large size regime (N ∼ 3000 atoms) show that the energy dispersion between the most stable elongated and isotropic particles is strongly sensitive to the nature of the metal–environment interaction and its strength. In extreme cases this may lead to stability crossovers between particles of different shapes and types. While such extreme cases were considered in order to make the discussed effects better pronounced, we note that the behavior of excess energy dispersion is continuous along the two considered series. As a consequence, the range of thermodynamically co-existing particle types increases progressively far before these extreme conditions are reached. Similarly, since nucleation barriers are proportional to excess energies, the shape diversity of critical nuclei is expected to increase following the progressive reduction of the excess energy dispersion, giving rise to an increasing diversity of growth shapes.It is worth reminding that the reported behavior of excess energy and shape dispersion could not have been obtained solely from the surface energy values, Fig. 2, using the Wulff construction. This evidently concerns the multi-twinned particles whose core is not bulk-like. It also applies whenever a stability inversion or quasi-degeneracy between isotropic and anisotropic particles takes place, as for MC NPs in environments (C) (Fig. 3) and (c) (Fig. 5). Finally, it is also the case of size-driven effects discussed in Section 5.
Two principal effects are to be held responsible for the trends in excess energy and shape reported in Section 4. On the one hand, unsurprisingly, the growing metal–environment interaction strength ε leads to an overall reduction of energies of facets and of more under-coordinated atoms (edges and corners). More interestingly, the ratio between these two energies changes in a different manner as a function of the environment nature (parameter p).
For the sake of simplicity, let us consider gold atoms of coordination numbers Zi = 9, 7, and 5, representative of the most dense (Zi = 9) and more open (Zi = 7) facets, and edges between them (Zi = 5), respectively. In analogy to the excess energy of NPs Δ, the atom formation energy with respect to bulk EB, γi = Ei − EB, represents the energy cost (per atom) of the corresponding facets and edges. Since in the case of gold the metal–metal contribution to γi is quasi-linear (Fig. 2, vacuum case), EM−Mi − EB ≈ k(ZB − Zi) (k ∼ 2.2 eV is the proportionality constant deduced from the linear dependence of vacuum surface energies as a function of the number of broken bonds in Fig. 2), and thus γi takes a simple analytical form:
| γi = k(ZB − Zi) − ε(ZB − Zi)p, | (5) |
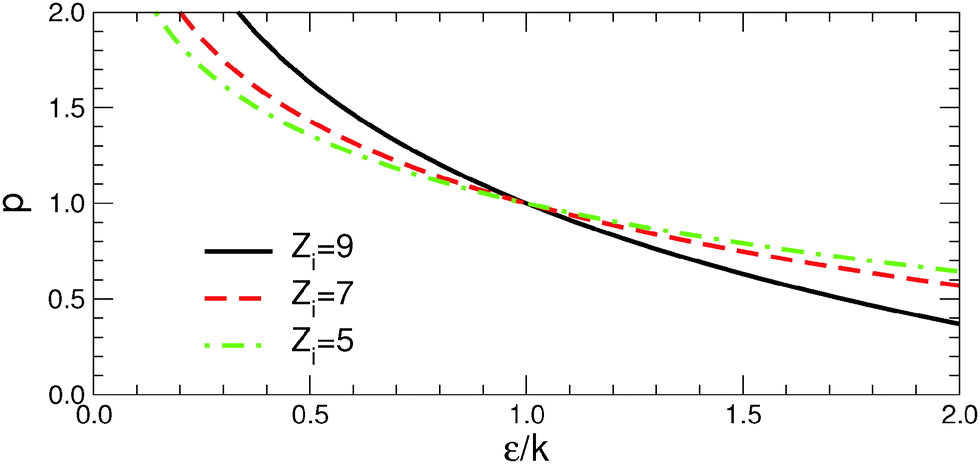 | ||
| Fig. 9 Critical lines in the diagram (ε/k, p), where the formation energies γi change sign for atoms of coordination numbers Zi = 9 (solid black line), 7 (dashed red line), and 5 (dash-dot green line) of eqn (5). See text for more details. | ||
More interestingly, Fig. 9 shows that if p > 1, the lower-coordinated atoms (e.g., those at the edges Zi = 5) are stabilized more efficiently than the higher-coordinated ones (e.g., those at the facets Zi = 7 and 9). In such environments, γedge/γfacet is a decreasing function of ε/k and, as a consequence, γedge vanishes or becomes negative before γfacet does. This is what happens along Series I, where the extreme environment (C) corresponds to the conditions where γfacet values are small but positive, while some γedge values become negative. This competition is the principal driving force for the stabilization of elongated particles. Conversely, in environments with p < 1, γedge/γfacet is an increasing function ε/k because higher-coordinated atoms are stabilized more efficiently than the lower-coordinated ones. The environment (d) of Series II, which belongs to the region of positive γi, is characterized by edge energies substantially larger than facet energies. Thus it does not produce a strong driving force for stabilization of anisotropic particles. Finally, we note that all critical lines intersect at a single point p = 1 and ε = k. In the particular case of pair-wise metal–environment interactions (p = 1), γedge/γfacet remains constant as a function of ε/k and the excess energies of all types of sites vanish at the same value of ε/k. The environment (c) of Series II, which is located close to this special point, is thus characterized by simultaneously the smallest facet and edge energies and provides the optimal conditions for an efficient reduction of the energy gap between particles of different shape and thus for stabilization of anisotropic NPs.
While changes in excess energies of MC and MT particles follow essentially similar trends as a function of (ε, p), the energy dispersion between these two types of particles changes along the two considered series. This comes from the fact that the shape of MT NPs is driven not only by the energy of facets and edges, but also by those of the twinning planes and the five-fold line defect. The latter do not depend explicitly on the environment, but their impact on the shape and stability of MT particles increases when the facet and edge energies become small. As a consequence, contrary to the vacuum conditions, where the critical size for stability inversion between decahedral (isotropic MT) and truncated octahedral (isotropic MC) particles is larger than 3000 atoms,32 in the considered environments, the MT particles are systematically less stable than the MC ones. However, the unfavorable effect of the MT particle core structure is small for rod-like shaped MT particles [e.g., environments (C) and (c)] due to a substantial reduction of the twinning plane size.
At small sizes (N < 1000 atoms), stability changes between isotropic and anisotropic NP shapes may have a different origin. In that size range, the optimal ratio of edge to surface atoms cannot be obtained whatever is the value of N, but rather at a discrete set of magic sizes only, as discussed already in the past for both unsupported and oxide-supported transition metal and noble clusters.37 This constraint gives rise to an oscillatory behavior of excess energies, Fig. 8, particularly well pronounced for the smallest particles, representative of the nucleation and very early growth stages. The same is true for strongly coordination-dependent environments, where a small change in N provokes a relatively important modification of the ratio of edge to surface atoms. In such cases, the difference in magic sizes between isotropic and elongated particles produces changes in their excess energy dispersion and their relative stability as a function of particle size. This gives a strong thermodynamic preference for the type and abundance of co-existing particles. Moreover, it suggests a possibility of tuning the initial shape of critical nuclei by the modification of nucleation size, via e.g., the supersaturation value.
Before concluding, we would like to point our that, very recently, the present model has successfully accounted for the existence of different types of elongated MT gold particles (either nanorods or bipyramids), synthesized in a CTAB solution in the presence or in the absence of silver(I), by an adequate choice of parameters ε and p.38
7 Conclusion
In this study, relying on a many-body potential approach for the description of transition or noble metal NPs interacting with an environment, we have sketched the behavior of energetics, shapes and morphologies of gold NPs as a function of the strength and nature of the metal–environment interaction. Thanks to the particular focus on two series of environments, we have highlighted conditions which enable a reduction of the energy difference between isotropic and elongated gold NPs, and which are thus propitious for the thermodynamic co-existence of these two types of NPs. The results of atomistic simulations on monocrystalline and multi-twinned particles have been rationalized with arguments involving the dependence of facet and edge energies on the environment.We have shown that increasing the strength of the metal–environment interaction always leads to a reduction of surface and edge energies, thus bringing the relative stability of large isotropic and anisotropic NPs closer to one another. The most favorable situation was found in the case of pair-wise metal–environment interactions, characterized by simultaneously small facet and edge energies and providing the optimal conditions for an efficient reduction of the energy gap between particles of different shapes. Conversely, in strongly coordination-dependent and covalent-like environments the ratio between surface and edge energies changes as the interaction strength increases. Especially in the latter case, this results in a much less pronounced bias for stabilization of anisotropic particles.
We have found that in most considered environments, the multi-twinned particles are systematically less stable than the monocrystalline ones, because the destabilizing effect of twinning planes becomes more pronounced upon reduction of the surface and edge energies. However, since this unfavorable effect is small in rod-like particles, the overall stability of elongated MT particles improves with an increase in metal–environment interaction strength.
For smaller particles, we have highlighted an oscillatory behavior of the energy differences between isotropic and anisotropic NPs as a function of particle size. Assigned to the different magic sizes of different types of particles, it is particularly important in strongly coordination-dependent environments, and gives a strong thermodynamic bias for a co-existence of the two types of particles at the nucleation and early growth stages.
Our results show that, by determining the abundance and diversity of particles nucleated in solution, thermodynamics may constitute an important force influencing their final shape. They provide firm grounds for simulations of particle growth and discussion of kinetic factors which influence particle shapes.
Acknowledgements
The authors acknowledge F. Testard, O. Spalla, L. Belloni, B. Fleury and C. Canbek for fruitful discussions, and financial support from the French project ANR-11-BS10-006 MIGRANI.References
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, R829 CrossRef CAS.
- A. M. Alkilany, S. E. Lohse and C. J. Murphy, Acc. Chem. Res., 2013, 46, 650–661 CrossRef CAS PubMed.
- A. Chen and S. Chatterjee, Chem. Soc. Rev., 2013, 42, 5425–5438 RSC.
- J. Pérez-Juste, I. Pastoriza-Santos, L. M. Liz-Marzán and P. Mulvaney, Coord. Chem. Rev., 2005, 249, 1870 CrossRef PubMed.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- B. Abécassis, F. Testard, O. Spalla and P. Barboux, Nano Lett., 2007, 7, 1723–1727 CrossRef PubMed.
- T. K. Sau and C. J. Murphy, Langmuir, 2004, 20, 6414 CrossRef CAS PubMed.
- F. Hubert, F. Testard and O. Spalla, Langmuir, 2008, 24, 9219 CrossRef CAS PubMed.
- Y. Takenaka and H. Kitahata, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 80, 020601 CrossRef.
- E. Carbó-Argibay, B. Rodríguez-González, S. Gómez-Graña, A. Guerrero-Martínez, I. Pastoriza-Santos, J. Pérez-Juste and L. M. Liz-Marzán, Angew. Chem., Int. Ed., 2010, 49, 9397–9400 CrossRef PubMed.
- F. Hubert, F. Testard, G. Rizza and O. Spalla, Langmuir, 2010, 26, 6887 CrossRef CAS PubMed.
- F. Hubert, F. Testard, A. Thill, Q. Kong, O. Tache and O. Spalla, Cryst. Growth Des., 2012, 12, 1548–1555 CAS.
- C. S. Ah, Y. J. Yun, H. J. Park, W.-J. Kim, D. H. Ha and W. S. Yun, Chem. Mater., 2005, 17, 5558–5561 CrossRef CAS.
- A. Halder and N. Ravishankar, Adv. Mater., 2007, 19, 1854–1858 CrossRef CAS.
- G. Cao and Y. Wang, Nanostructures and Nanomaterials: Synthesis, Properties and Applications, World Scientific, 2nd edn, 2011, vol. 2 Search PubMed.
- A. Halder, P. Kundu, B. Viswanath and N. Ravishankar, J. Mater. Chem., 2010, 20, 4763–4772 RSC.
- G. Grochola, S. P. Russo and I. K. Snook, J. Chem. Phys., 2007, 126, 164707 CrossRef PubMed.
- G. D. Barmparis and I. N. Remediakis, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 085457 CrossRef.
- K. P. McKenna and A. L. Shluger, J. Phys. Chem. C, 2007, 111, 18848–18852 CAS.
- K. P. McKenna, Phys. Chem. Chem. Phys., 2009, 11, 4145–4151 RSC.
- G. D. Barmparis, K. Honkala and I. N. Remediakis, J. Chem. Phys., 2013, 138, 064702 CrossRef PubMed.
- S. K. Meena and M. Sulpizi, Langmuir, 2013, 29, 14954–14961 CrossRef CAS PubMed.
- S. E. Lohse, N. D. Burrows, L. Scarabelli, L. M. Liz-Marzan and C. J. Murphy, Chem. Mater., 2014, 26, 34–43 CrossRef CAS.
- N. Almora-Barrios, G. Novell-Leruth, P. Whiting, L. M. Liz-Marzán and N. López, Nano Lett., 2014, 14, 871–875 CrossRef CAS PubMed.
- R. Cortes-Huerto, J. Goniakowski and C. Noguera, J. Chem. Phys., 2013, 138, 244706 CrossRef PubMed.
- D. Pettifor, Bonding and Structure of Molecules and Solids, Oxford University Press, 1995 Search PubMed.
- M. Finnis, Interatomic Forces in Condensed Matter, Oxford University Press, 2003 Search PubMed.
- F. Cyrot-Lackmann and F. Ducastelle, Phys. Rev. B: Solid State, 1971, 4, 2406–2412 CrossRef.
- R. P. Gupta, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 6265–6270 CrossRef CAS.
- V. Rosato, B. Guillopé and B. Legrand, Philos. Mag. A, 1989, 59, 321 CrossRef.
- H. Chamati and N. I. Papanicolaou, J. Phys.: Condens. Matter, 2004, 16, 8399 CrossRef CAS.
- B. Hammer and J. Nørskov, Impact of Surface Science on Catalysis, Academic Press, 2000, vol. 45, pp. 71–129 Search PubMed.
- M. C. Desjonquères and D. Spanjaard, Concepts in Surface Physics, Springer Verlag, 1993 Search PubMed.
- F. Baletto, R. Ferrando, A. Fortunelli, F. Montalenti and C. Mottet, J. Chem. Phys., 2002, 116, 3856 CrossRef CAS PubMed.
- J. Goniakowski and C. Mottet, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155443 CrossRef.
- C. Mottet, J. Goniakowski, F. Baletto, R. Ferrando and G. Treglia, Phase Transitions, 2004, 77, 101–113 CrossRef CAS.
- Z. C. Canbek, R. Cortes-Huerto, F. Testard, O. Spalla, S. Moldovan, O. Ernsen, A. Wisnet, G. Wang, J. Goniakowski, C. Noguera and N. Menguy, submitted.
Footnote |
| † Present address: Max-Planck-Institut für Polymerforschung, Ackermannweg 10, 55128 Mainz, Germany. |
| This journal is © the Owner Societies 2015 |