
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH-Assisted control over the binding and relocation of an acridine guest between a macrocyclic nanocarrier and natural DNA†
Mhejabeen
Sayed
* and
Haridas
Pal
*
Radiation & Photochemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400 085, India. E-mail: msayed@barc.gov.in; hpal@barc.gov.in; Fax: +91-22-25505151
First published on 26th February 2015
Abstract
The differential binding affinity of the hydroxypropyl-β-cyclodextrin (HPβCD) macrocycle, a drug delivery vehicle, towards the protonated and deprotonated forms of the well-known DNA binder and model anticancer drug acridine has been exploited as a strategy for dye–drug transportation and pH-responsive delivery to a natural DNA target. From pH-sensitive changes in the ground state absorption and steady-state fluorescence characteristics of the studied acridine dye–HPβCD–DNA ternary system and strongly supported by fluorescence lifetime, fluorescence anisotropy, Job's plots, 1H NMR and circular dichroism results, it is revealed that in a moderately alkaline solution (pH ∼ 8.5), the dye can be predominantly bound to the HPβCD macrocycle and when the pH is lowered to a moderately acidic region (pH ∼ 4), the dye efficiently detaches from the HPβCD cavity and almost exclusively binds to DNA. In the present study we are thus able to construct a pH-sensitive supramolecular assembly where pH acts as a simple stimulus for controlled uptake and targeted release of the dye–drug. As pH is an essential and sensitive factor in various biological processes, a simple yet reliable pH-sensitive model such as is demonstrated here can have promising applications in the host-assisted delivery of prodrug to the target sites, such as cancer or tumour microenvironments, with an enhanced stability, bioavailability and activity, and also in the design of new fluorescent probes, sensors and smart materials for applications in nano-science.
Introduction
Supramolecular assemblies formed with the involvement of non-covalent interactions have recently attracted immense research interest due to their diverse utility in areas such as drug delivery,1,2 nanotechnology,3 the food industry,4 on–off switches,5 catalysis,6 photostabilization,2,7 photodynamic therapy,8 optical sensors,9 and many others.10,11 The underlying non-covalent host–guest interactions have great potential for developing excellent tunable functional materials because of their strong responses to external stimuli such as pH,1,12–15 salt,11,15 temperature,5,15 light,5etc. Stimuli-responsive targeted drug delivery, particularly involving macrocyclic hosts as carriers, is currently undergoing great advances in pH, salt or light acting as triggers for controlled cargo drug release at the target site for its desired effects with enhanced efficiency.2,5,11–15 In supramolecular drug delivery applications, the drug carrier vehicle plays a vital role in transporting the drug specifically to the required location and thus increasing the concentration and also the effectiveness of the drug at the targeted site, which in turn reduces the toxicity and undesired side effects to normal cells.1,2,13–16 Cyclodextrins (CDs) as macrocyclic hosts have been very successfully used as drug carriers in many drug formulations.1,16,17CDs are unique cyclic polysaccharides formed from D-glucopyranose monomer units joined by ether linkages in a cyclic manner and thus provide hydrophobic cavities where nonpolar/hydrophobic residues of the guest molecules can be encapsulated to form inclusion complexes via non-covalent interactions.7,13,18–21 Depending upon the number of D-glucopyranose units present, different CD homologues with varying cavity sizes exist, namely αCD, βCD and γCD, containing 6, 7, and 8 monomer units, respectively. Cyclodextrins and their derivatives show low toxicity, high biocompatibility and excellent inclusion capability with a variety of drug–dye molecules, which make them attractive for uses in several drug formulations and many biomedical applications.1,2,7,8,13–17 In this regard CD derivatives with polar substituents are more promising than the parent CD hosts. For example, hydroxypropyl-β-cyclodextrin (HPβCD; Scheme 1) shows dramatically increased water solubility and much-enhanced binding interactions in comparison to parent CDs and thus can largely improve the bioavailability of the drugs. Moreover, like CDs, the HPCDs have also been reported to be well-tolerated in humans.1,22 Recently, Huang et al. have reported significantly improved water solubility, thermal stability, dissolution rate and increased sprout inhibition effect in potatoes for the drug chlorpropham on complexation with HPβCD.23 Miyake, et al. have also reported that the drug rutin shows increased bioavailability in a formulation made with HPβCD compared to that made with βCD.24
 | ||
| Scheme 1 The prototropic equilibrium of acridine dye and the HPβCD host used in this study are shown for quick visualization. | ||
DNA is considered to be one of the primary intracellular targets for anticancer drugs, where the drug can cause selective damage of cancer cells,25–27 taking advantage of the acidic pH conditions of the cancerous organelles.13,28 Acridine dye and its derivatives are widely used in antitumor treatment as well as in chemotherapy. Many new acridine derivatives are being prepared, improving their DNA binding properties to give superior antitumor activity.26,29–31 Selective delivery of such anticancer drugs at target sites that have low pH, like tumour cells, may increase the localized cytotoxicity and lower the side effects of the drug towards healthy cells.
In the present study we have investigated the interaction of the model anticancer drug acridine with HPβCD and a biomolecular target DNA at different pH conditions in aqueous solution, to find out if a pH-responsive release of the model drug to the target can be achieved. Acridine is a prototropic dye (cf.Scheme 1), with a pKa of 5.4.19 Thus, in aqueous solution it exists in the protonated form (AcH+) at acidic pH and in neutral form (Ac) at alkaline pH.19 Acridine and its derivatives are potent DNA binders because of their planar structures,29,30,32 and are well-recognized as drugs showing carcinogenic effect due to their property to bind strongly with DNA. Acridine dyes are reported to exhibit two modes of binding with DNA: intercalative binding, where the planar ring of the acridines intercalates into the DNA base pairs, and semi-intercalative or exo-binding, where electrostatic interaction with the phosphate backbone of DNA plays a major role.33–35 Significant microenvironment-sensitive changes in the spectroscopic properties of acridine dyes make them useful fluorescence probes for studying local environments in various microheterogeneous systems.36–38 Taking advantage of the strikingly different binding behaviour of the HPβCD macrocycle towards the protonated and neutral forms of acridine dye, as observed in the present study, we have demonstrated an effective pH-triggered controlled and targeted release of the dye from the HPβCD cavity, a potential drug carrier vehicle, to the target site of DNA, which is a major goal in chemotherapeutic applications. Though the interaction of organic dyes with cyclodextrins and DNA has been reported in the literature,39,40 detailed investigations into pH response in such supramolecular assemblies are very limited. In contrast to involved multi-step synthesis and sophisticated detection methods, we present here a simple yet reliable pH-responsive model study using photochemical measurements, which can open up new opportunities in engineering intelligent functional materials useful for various biomedical applications.
Results and discussion
1.1. Interaction of acridine dye with DNA and HPβCD hosts
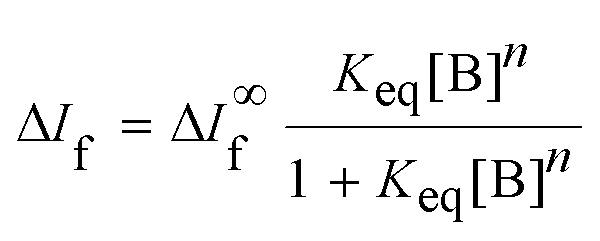 | (1) |
 | ||
| Fig. 1 (a) Changes in the SS fluorescence spectra for the AcH+–DNA system at pH 4; [AcH+] = 10.2 μM and [DNA] = 0, 36, 59, 78, 117, 172, 244, 296, 330, 396, 429, 522, 694 and 800 μM for spectra 1–14. Inset: fluorescence titration curve for the AcH+–DNA system at pH 4, analysed using eqn (1) with n = 2. (b) Changes in the SS fluorescence spectra for the Ac–DNA system at pH 8.5; [Ac] = 10.5 μM and [DNA] = 0, 10, 30, 69, 135, 209, 279, 364, 522, 667 and 800 μM for spectra 1–11. Inset: fluorescence titration curve for the Ac–DNA system at pH 8.5, analysed using eqn (1) with n = 2. | ||
The ground state absorption behaviour was also examined for both the AcH+ and Ac forms of the dye in the presence of DNA. The absorption maximum for both forms of the dye appears at about 355 nm. The AcH+ form is, however, distinctly characterized by its broad longer-wavelength shoulder absorption band spreading into the 380–440 nm region, which is absent for the neutral form of the dye.19 Upon gradual addition of DNA to the AcH+ and Ac solutions (at pH 4 and 8.5, respectively), the peak absorbance shows a gradual decrease along with small bathochromic shifts (∼1 nm for AcH+ and ∼3 nm for Ac for the 354 nm band) and the appearance of isosbestic point-like features at ∼360 nm for AcH+ and ∼392 nm for Ac (cf. Fig. S1 in the ESI†). These results are thus in corroboration with the SS fluorescence results presented in Fig. 1a and b suggesting a strong binding interaction for both prototropic forms of the dye with the DNA host.
The n value of 2 obtained from the analysis of the fluorescence titration data for both AcH+–DNA and Ac–DNA systems indicates that there is simultaneous involvement of two nucleotides in the binding of a dye molecule to the DNA host. To verify such a stoichiometry further for the host–guest complexes in the AcH+–DNA and Ac–DNA systems, we carried out Job's plot measurements42,43 following both absorption and fluorescence studies. Fig. 2 shows the Job's plots for the AcH+–DNA (pH 4) and Ac–DNA (pH 8.5) systems, respectively, obtained from fluorescence measurements. The Job's plots for these systems as obtained from absorption measurements are shown in Fig. S2 in the ESI.† The maxima of these Job's plots appear at around a 0.33 mole fraction of the dye and thus unambiguously establish the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (dye to host) stoichiometry of the complexes formed in both the AcH+–DNA and Ac–DNA systems.
:2 (dye to host) stoichiometry of the complexes formed in both the AcH+–DNA and Ac–DNA systems.
 | ||
| Fig. 2 Job's plots for the AcH+–DNA (pH 4) and Ac–DNA (pH 8.5) systems obtained from fluorescence changes (ΔIf = Idye–host − Idye-only) as a function of the constituents. The sum of the dye and the host concentrations in these measurements was kept constant at 50 μM. The respective excitation and emission wavelengths were 360 nm and 482 nm for the AcH+–DNA system, and 361 nm and 430 nm for the Ac–DNA system. | ||
Knowing about the strong interaction of the dye with DNA, we subsequently carried out absorption and SS fluorescence measurements to explore the interaction of the dye with the recognized drug carrier, HPβCD. For the Ac form of the dye (pH 8.5), there is a gradual decrease in the fluorescence intensity with an increase in the HPβCD concentration, along with the concomitant development of two new vibrational features in the spectra at ∼427 and 445 nm and also a small hypsochromic shift in the emission maxima, as shown in Fig. 3. These results indicate formation of quite a strong inclusion complex for the Ac form of the dye with the HPβCD host. The hypsochromic shift and the development of vibrational features clearly suggest that the bound dye experiences a lower micropolarity on incorporation into the HPβCD cavity than in bulk water. Reduction in the fluorescence intensity for the Ac–HPβCD systems is justifiably assigned to the strong hydrogen bonding interaction of the encapsulated Ac with the portal OH groups of the HPβCD host, as was also observed in our earlier study on the interaction of the dye with the unsubstituted βCD host.19
 | ||
| Fig. 3 Changes in the SS fluorescence spectra for the Ac–HPβCD system at pH 8.5; [Ac] = 13.6 μM and [HPβCD] = 0, 0.2, 0.43, 0.5, 0.81, 1.85, 3.9, 6.6, and 9.1 mM for spectra 1–9. Inset: fluorescence titration curve for the Ac–HPβCD system at pH 8.5, analysed using eqn (1) with n = 1. | ||
In accordance with the SS fluorescence results, in the absorption studies also there is a small decrease in the absorbance along with a small red shift (∼2 nm) in the absorption maximum for the Ac form (pH 8.5) of the dye on addition of the HPβCD host in the solution (cf. Fig. S3a, ESI†). Observed results support a reasonable host–guest interaction in the Ac–HPβCD system. Unlike the neutral Ac form, the protonated AcH+ form of the dye, however, did not show any appreciable change, either in the absorption (cf. Fig. S3b, ESI†) or in the fluorescence (cf. Fig. S4, ESI†) characteristics, on addition of the HPβCD host in the solution. These observations suggest that the interaction of the AcH+ form of the dye with HPβCD host is extremely weak in comparison to that of the neutral Ac form of the dye.
A Job's plot study42,43 was carried out for the Ac–HPβCD system at pH 8.5 to explore the stoichiometry of the complex formed in this case. Due to very small changes that occur in the absorbance of Ac on addition of the HPβCD host (cf. Fig. S3a, ESI†), no meaningful Job's plot could be obtained for the Ac–HPβCD system from absorption measurements. A satisfactory Job's plot for the present system, however, could be obtained from the fluorescence study, as shown in Fig. 4, indicating a maximum at around a 0.5 mole fraction of the dye and thus suggesting the 1:1 stoichiometry of the complex formed in this system. For the AcH+–HPβCD (pH 4) system, as the interaction was found to be extremely weak from both absorption and fluorescence studies (cf. Fig. S3b and S4, ESI†), a Job's plot measurement was not attempted for this system.
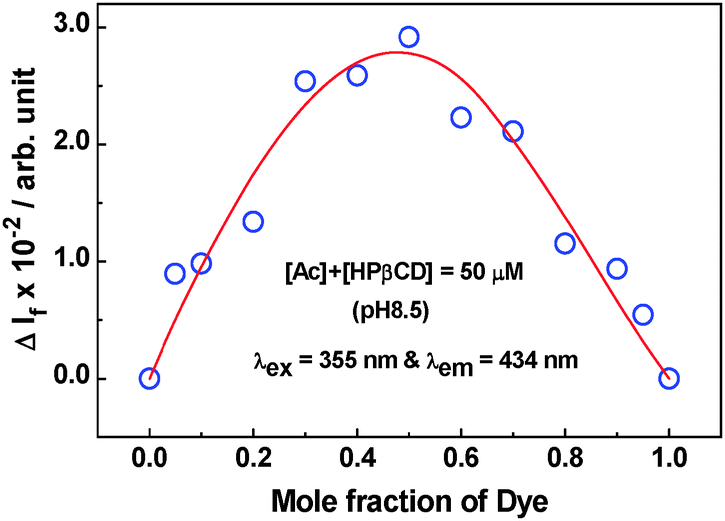 | ||
| Fig. 4 Job's plot obtained for the Ac–HPβCD (pH 8.5) system using fluorescence measurement. The sum of the dye and the host concentrations in these measurements was kept constant at 50 μM. Excitation and emission wavelengths were 355 nm and 434 nm, respectively. | ||
A fluorescence titration method was utilized to evaluate the binding constant (Keq) value for the Ac–HPβCD system. As shown in the inset of Fig. 3, the changes in the fluorescence intensity (ΔIf) as a function of the host concentration fit well with the consideration of 1:1 complex formation (cf.eqn (1) with n = 1), as suggested from the Job's plot. The Keq value thus estimated for the Ac–HPβCD complex is about 1.0 × 103 M−1. It should be mentioned here that the binding interaction of Ac with HPβCD is about 3.4 times higher than that observed with the parent βCD host, reported in our earlier study.19 It is thus evident that the extended cage structure offered by the HPβCD host renders a much greater hydrophobic interaction for the Ac form of the dye, leading to formation of a stronger inclusion complex. For the AcH+ form of the dye, as there was no appreciable change either in the absorption or in fluorescence characteristics (cf. Fig. S3b and S4, ESI†), no titration study could be carried out for the AcH+–HPβCD system to estimate the binding constant value.
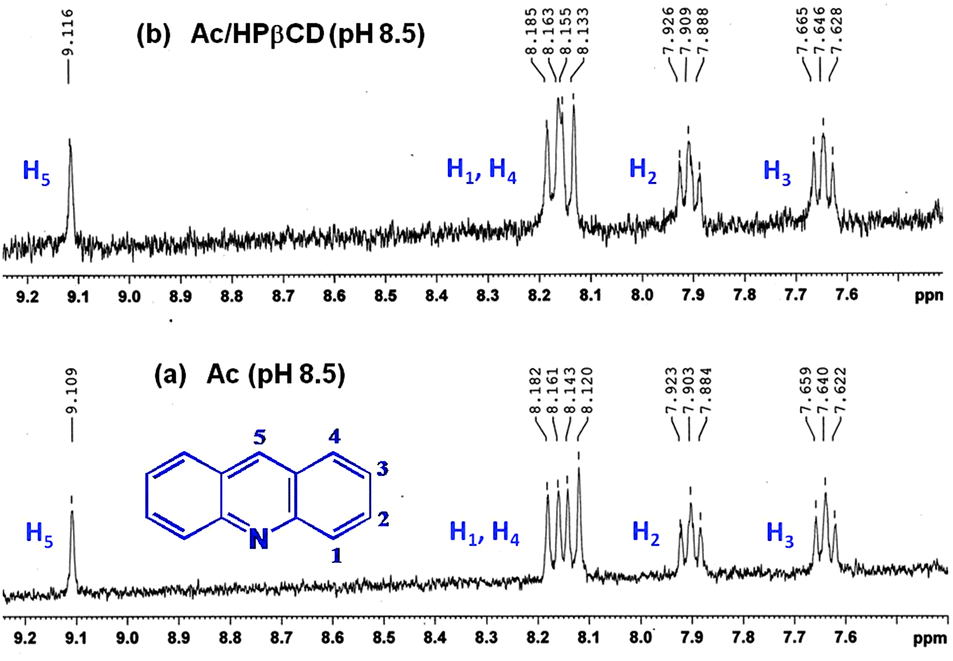 | ||
| Fig. 5 1H NMR spectra of (a) free Ac and (b) the Ac–HPβCD system obtained at pH 8.5. Concentrations of the components were: [Ac] = 150 μM and [HPβCD] = 150 mM. | ||
1H NMR studies were also carried out for the Ac–DNA system in D2O solution at pD 8.5. The NMR spectra recorded for the free Ac and the Ac–DNA system under similar experimental conditions are shown in Fig. S5 of the ESI.† As indicated from this figure, there are no distinguishable NMR signals for the Ac–DNA system that can be attributed either to the guest dye or to the DNA host (cf. Fig. S5b, ESI†), even though characteristic NMR signals are clearly observed for free dye in the absence of DNA under similar experimental conditions (cf. Fig. S5a, ESI†). As the DNA molecules possess a large number of exchangeable protons, it appears that in the present experimental conditions all the proton signals of DNA are exceedingly well-broadened causing the development of a significant background such that not only the DNA signals but also those of the guest dye are masked under this background. As the NMR results for the Ac–DNA system were not conclusive, a similar NMR study for the AcH+–DNA system was not carried out in the present work.
 | ||
| Scheme 2 Four-state thermodynamic equilibrium model for the acridine dye and host (H, either HPβCD or DNA) systems considering all the stages of host–guest interaction and the acid dissociation processes. | ||
The modulation of the acid–base properties of the dye upon its interaction with DNA and HPβCD hosts was investigated systematically by following the pH-dependent changes in the absorption spectra of the dye in the presence of significantly high concentrations of the hosts, as shown in Fig. S6 of the ESI.† Absorbance changes were noted at selected wavelengths in the absence as well as in the presence of DNA and HPβCD hosts, separately, as a function of the pH of the solution, as shown in Fig. 6. The pKa values were estimated from the inflection points of the plots and are found to be 5.4, 5.0 and 4.4 for the free dye, dye–DNA and dye–HPβCD systems, respectively.
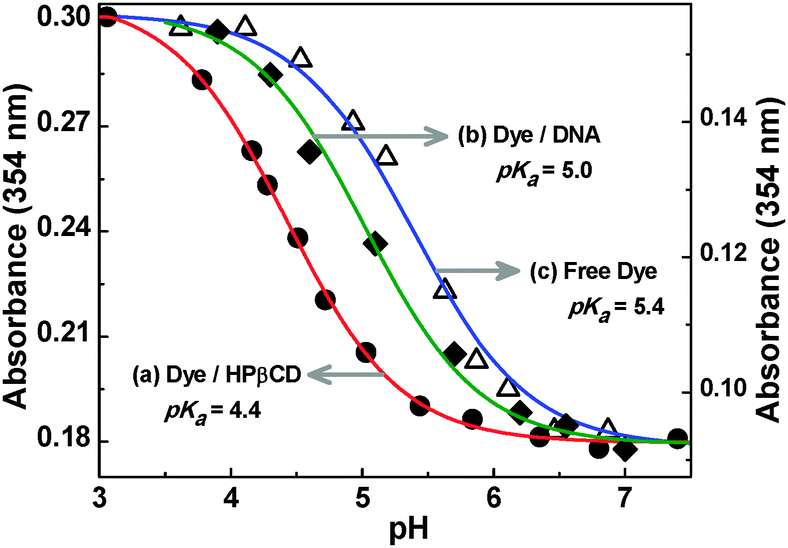 | ||
| Fig. 6 Changes in the absorbance of acridine dye (10 μM) at 354 nm measured in the presence of (a) 30 mM HPβCD, (b) 800 μM DNA and (c) in the absence of any host, as a function of the pH of the solution. | ||
As indicated by the observed results, the pKa value of the dye undergoes about 0.4 and 1.0 units of downward shift on its interaction with DNA and the HPβCD hosts, respectively, which supports the preferential binding of the Ac form of the dye with both the DNA and HPβCD hosts compared to the AcH+ form, as inferred earlier from the fluorescence titration studies (cf. Section 1.1.1). The larger pKa shift for the dye–HPβCD system (1.0 units) compared to that of the dye–DNA system (0.4 units) is directly in accordance with the observation that there is a larger difference in the binding affinities for the Ac and AcH+ forms of the dye with HPβCD host than with DNA. Importantly, the difference in the pKa shifts between the dye–DNA and dye–HPβCD systems can be exploited suitably to achieve a preferential binding for the dye, either to the DNA or to the HPβCD host, in a given dye–DNA–HPβCD ternary system, just by adjusting the pH of the solution, as discussed in the following sections.
1.2. Competitive interaction of acridine dye with HPβCD and DNA hosts at pH 5.5
Competitive binding interaction of the two prototropic forms of acridine dye has been investigated at pH 5.5, a characteristic pH of cancer cells, in the simultaneous presence of both DNA and HPβCD hosts. As expected, at this pH the absorption spectrum of the free dye (cf.Fig. 7) exhibits a broad absorption shoulder at the longer wavelength region (∼400–440), an exclusive feature for the AcH+ form of the dye, along with the absorption maximum at 354 nm. These observations are consistent with the pKa value of 5.4 for the dye,19 suggesting the presence of both AcH+ and Ac forms of the dye in the solution at pH 5.5, both with significant proportions (almost 50:50).
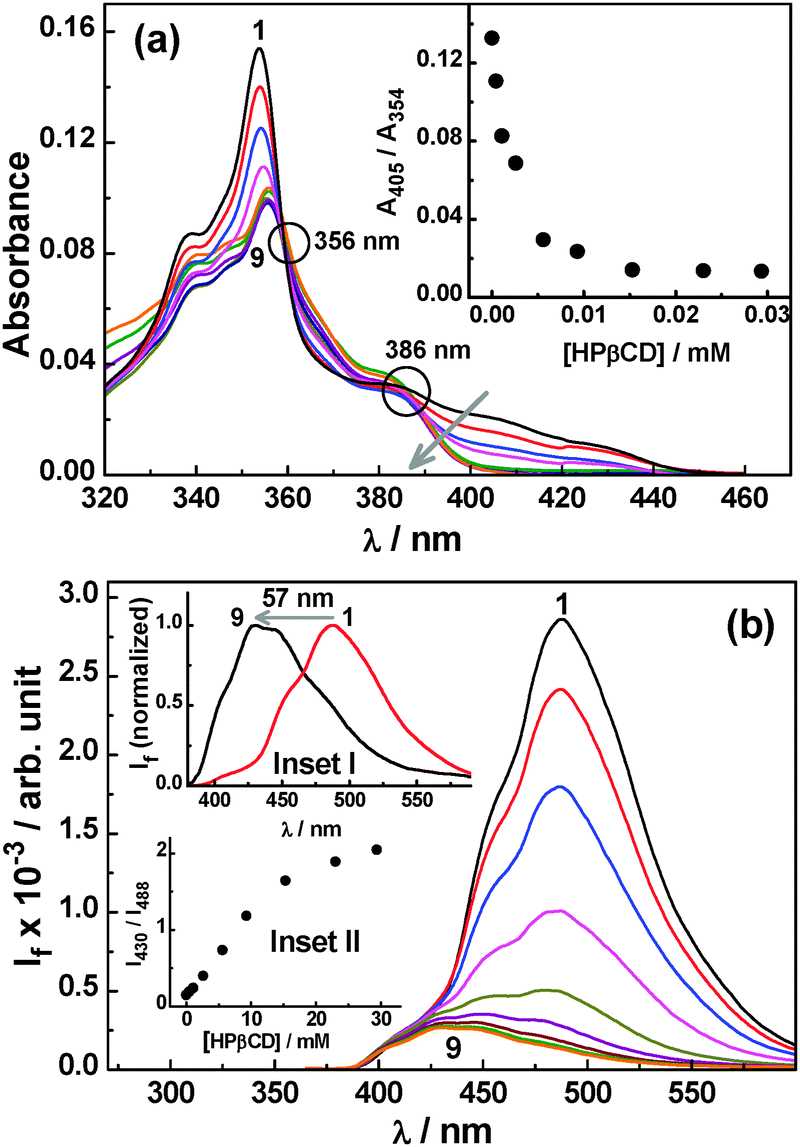 | ||
| Fig. 7 (a) Changes in the absorption spectra for the Ac–HPβCD system at pH 5.5; [Ac] = 10 μM and [HPβCD] = 0, 0.46, 1.08, 2.6, 5.6, 9.3, 15.3, 23, and 29.4 mM for spectra 1–9. Inset: ratio of absorbance at 405 and 354 nm (A405/A354) as a function of HPβCD concentration. (b) Changes in the SS fluorescence spectra for the Ac–HPβCD system at pH 5.5; [Ac] = 10.6 μM and [HPβCD] = 0, 0.46, 1.08, 2.6, 5.6, 9.3, 15.3, 23, and 29.4 mM for spectra 1–9. Inset I: normalized fluorescence spectra for (1) only Ac and (9) Ac–HPβCD (29.4 mM) at pH 5.5. Inset II: ratio of fluorescence intensities at 430 and 488 nm (I430/I488) as a function of HPβCD concentration. | ||
Fig. 7a shows the changes in the absorption spectra of the dye with changing HPβCD concentration at pH 5.5. With an increase in HPβCD concentration, there is a decrease in the absorbance for both the 354 nm main band and the 400–440 nm shoulder band along with a small red shift in the main absorption band by ∼3 nm. These changes are certainly due to the formation of the dye–HPβCD inclusion complex. Interestingly, it is observed from Fig. 7a that in the presence of HPβCD at pH 5.5 there is a relatively larger decrease in the absorbance for the 400–440 nm shoulder band in comparison to that of the 354 nm main band. This observation suggests that the relative proportion of the Ac form is enhanced in the solution upon increasing the HPβCD concentration, as a consequence of the downward pKa shift of the dye due to preferential Ac–HPβCD complex formation. Appearance of the kind of isosbestic points at about 360 and 386 nm in the absorption spectra with increasing HPβCD concentration also supports the conversion of some of the AcH+ to the Ac form through the preferential formation of the Ac–HPβCD complex. This is further indicated by the plot shown in the inset of Fig. 7a, displaying a sharp decrease in the ratio of absorbance at 405 nm (mainly due to AcH+) to that at 354 nm (both AcH+ and Ac contributes) with an increase in the HPβCD concentration.
Studies on the acridine dye–HPβCD system at pH 5.5 have also been carried out using SS fluorescence measurements and the results are shown in Fig. 7b. It is observed that upon increasing the HPβCD concentration there is a large decrease in the fluorescence intensity along with a large blue shift in the emission peak, as large as ∼57 nm (cf. inset I of Fig. 7b). These results clearly manifest the switch over of a large fraction of the AcH+ emission in the absence of HPβCD to the predominantly Ac emission in the presence of HPβCD, suggesting the host-assisted deprotonation of AcH+ at the studied pH conditions. It is thus evident that, without changing the pH, one can change the proportions of the prototropic forms of the dye in the solution, just by the addition of the HPβCD host. Inset II of Fig. 7b, shows the plot of fluorescence intensity ratio (I430/I488) for the dye at 430 nm (Ac) to 488 nm (AcH+), as a function of the increasing HPβCD concentration. As revealed from this plot, the intensity ratio increases sharply upon increasing the host concentration, indicating the preferential formation of the Ac–HPβCD inclusion complex in the system and thereby a shift in the overall prototropic equilibrium of the dye gradually towards the Ac form. The Keq value for the Ac–HPβCD system was also estimated from fluorescence titration data at pH 5.5 (cf. Fig. S7a, ESI†) and is found to be 560 M−1, half of the value estimated at pH 8.5. This reduction in the Keq value arises because at pH 5.5 a fraction of dye remains in the protonated form which does not interact with HPβCD (cf. Section 1.1.1).
As discussed in Section 1.1, both HPβCD and DNA show greatly different binding affinities for the AcH+ and Ac forms of the dye. Such a property can be exploited to control the binding of the dye preferentially to either the HPβCD or DNA hosts, simply by adjusting the pH of the solution. To realize such control and to achieve a possible dye relocalization from HPβCD to DNA, the photochemical changes for the dye (10 μM)/HPβCD (30 mM) system at pH 5.5 was further explored in the presence of varying DNA concentrations. Fig. 8a depicts the changes in the absorption spectra of the Ac–HPβCD system with changing DNA concentration. The results show an appreciable regain of the broad longer wavelength shoulder band of the AcH+ form of the dye, implying that the dye slowly detaches from the Ac–HPβCD complex and subsequently binds to the DNA host through protonation of the detached dye. This is further evidenced from the appearance of the clear isosbestic point at ∼390 nm. Formation of a good extent of the AcH+–DNA complex is also supported by the comparison of the absorption spectra in the related binary and ternary systems (cf. inset I of Fig. 8a) and by the changes in the absorbance ratio of 415 nm to 356 nm for the ternary system (cf. inset II of Fig. 8a). The observed results clearly indicate that at pH 5.5, the dye prefers to bind to the DNA host over HPβCD through the formation of the AcH+–DNA complex in lieu of the Ac–HPβCD complex. This is in accordance with the higher pKa value (5.0) for the dye–DNA system as compared to that of the dye–HPβCD system (4.4).
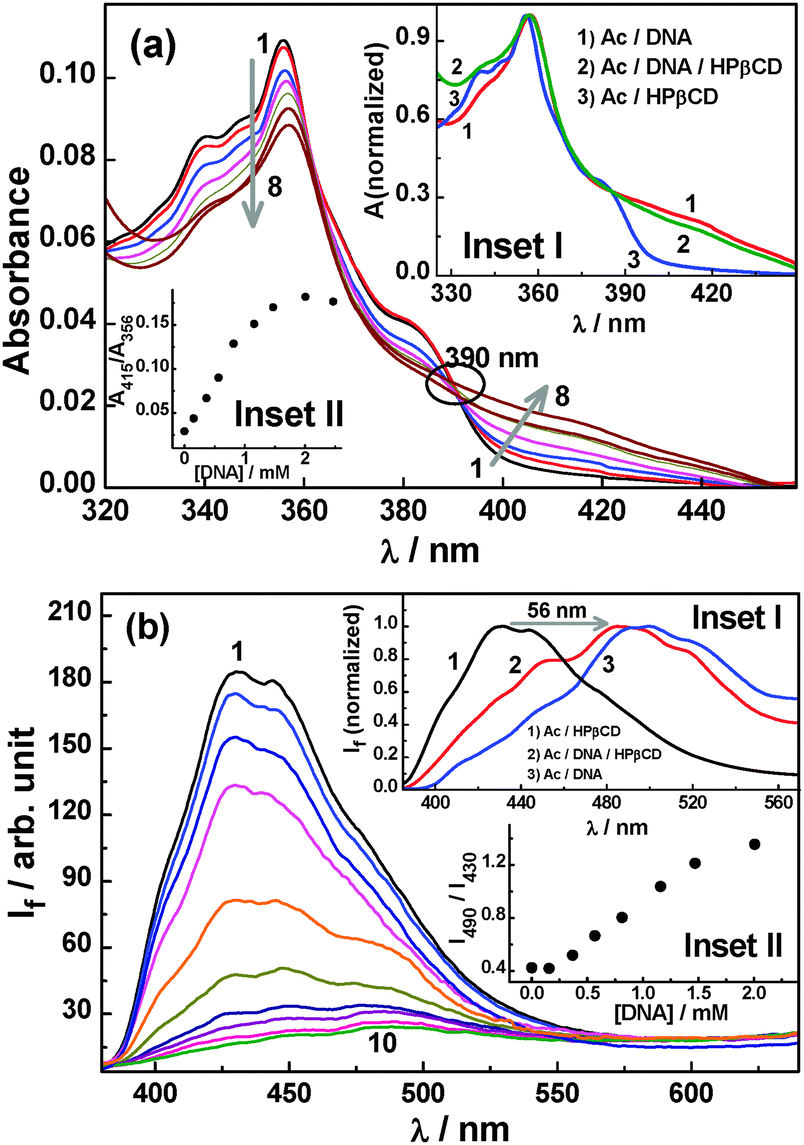 | ||
| Fig. 8 (a) Changes in the absorption spectra for the Ac–HPβCD system upon addition of DNA at pH 5.5; [Ac] = 11 μM, [HPβCD] = 30 mM and [DNA] = 0, 0.16, 0.37, 0.57, 0.81, 1.16, 2.02, and 2.47 mM for spectra 1–8. Inset I: normalized absorption spectra for (1) Ac–DNA, (2) Ac–DNA–HPβCD and (3) Ac–HPβCD at pH 5.5; [Ac] = 11 μM, [HPβCD] = 30 mM and [DNA] = 2.47 mM. Inset II: ratio of absorbances at 415 and 356 nm with increasing DNA concentration. (b) Changes in the SS fluorescence spectra for the Ac–HPβCD system upon addition of DNA at pH 5.5; [Ac] = 11 μM, [HPβCD] = 30 mM and [DNA] = 0, 0.07, 0.16, 0.37, 0.57, 0.81, 1.16, 1.47, 2.02, and 2.47 mM for spectra 1–10. Inset I: normalized fluorescence spectra for (1) Ac–HPβCD, (2) Ac–DNA–HPβCD and (3) Ac–DNA at pH 5.5. [Ac] = 11 μM, [HPβCD] = 30 mM and DNA = 2.47 mM. Inset II: ratio of fluorescence intensities at 490 and 430 nm with increasing DNA concentration. | ||
The fluorescence spectral features of the Ac–HPβCD system at pH 5.5 also undergo large changes upon the addition of DNA, as shown in Fig. 8b. With an increase in the DNA concentration, the fluorescence intensity undergoes a large reduction, indicating the disruption of the dye–HPβCD complex in the presence of DNA. Additionally, the fluorescence peak at 430 nm slowly disappears and a prominent new peak appears at 487 nm, illustrating the substantial formation of the AcH+–DNA complex in the system. In the present case, since the solution pH is close to the dye pKa, a reasonable amount of the Ac–HPβCD and Ac–DNA complexes are present in the solution, as indicated by the small hump at around 450 nm for the Ac–DNA–HPβCD system (cf. inset I of Fig. 8b). Upon comparison of the fluorescence spectra for the relevant binary and ternary systems, as shown in the inset I of Fig. 8b, it is clearly exemplified that DNA dominates the binding of the dye at pH 5.5 compared to the HPβCD host. It is noticed from inset II of Fig. 8b that the fluorescence intensity ratio for the acridine dye between 490 and 430 nm (I490/I430) increases reasonably sharply with an increase in the DNA concentration. This observation is directly in corroboration with the fact that at pH 5.5 the dye shows a preferential binding towards DNA over HPβCD, resulting in a major fraction of the dye undergoing relocation from the HPβCD nanocavity to the DNA pocket. Thus, at pH 5.5, the HPβCD in effect plays the role of a drug transport vehicle, supplying the dye–drug for binding to the DNA target. The binding constant value for the formation of the new AcH+–DNA complex in the presence of HPβCD was estimated following eqn (1) and is found to be 4.6 × 106 M−2, which is ∼a quarter of the value estimated at pH 4 in the absence of HPβCD (Fig. S7b, ESI†). This is expected because there is a competition between DNA and HPβCD to bind the dye and hence the dye binding with DNA is significantly affected by the presence of HPβCD and vice versa. In the present case, the number of nucleotides (n) required to bind a dye is also found to be two (cf. Fig. S7b, ESI†), similar to that obtained in the absence of the HPβCD host at pH 4 and 8.5, suggesting that the HPβCD does not have any direct interaction with DNA such that the mode of dye binding to DNA remains unaltered. It should be noted here that in the literature it is also well-documented that there is hardly any interaction of cyclodextrin hosts with DNA.45,46
1.3. pH triggered delivery of acridine dye from HPβCD host to DNA
As binding interactions in the studied systems are noncovalent in nature, it is interesting to see if the dye binding and relocation between the HPβCD and DNA can be controlled at will using pH as a stimulus, which could eventually be exploited rationally in applications such as controlled drug delivery. Taking into consideration the pH range where denaturation of DNA does not take place (pH 4 to 9),47–50 we systematically investigated the dye–HPβCD–DNA ternary system at two different pH conditions, namely pH 4 and 8.5, where acridine dye will exist mainly in the AcH+ and Ac forms, respectively. Results of these studies are very intriguing and are described in the following sections. , which is equal to ∼5 × 103 M−1. This value is about 5 times larger than the Keq value for the Ac–HPβCD system (∼1 × 103 M−1). Though the comparison of these binding constants suggests that the dye can reside better in DNA, the observed spectral features suggest that the dye is preferentially going into the HPβCD nanocavity under the present experimental conditions. The preferential binding of the dye to HPβCD is understandably assisted by about a five times higher concentration of HPβCD compared to DNA used in the solution. Thus, from the experimental results, it is very evident that in the dye–HPβCD–DNA ternary system at pH 8.5, the dye can be preferentially bound to the HPβCD cavity in its neutral form Ac, using a reasonably high concentration of the macrocyclic host and thereby resisting the binding of the dye to the DNA host.
, which is equal to ∼5 × 103 M−1. This value is about 5 times larger than the Keq value for the Ac–HPβCD system (∼1 × 103 M−1). Though the comparison of these binding constants suggests that the dye can reside better in DNA, the observed spectral features suggest that the dye is preferentially going into the HPβCD nanocavity under the present experimental conditions. The preferential binding of the dye to HPβCD is understandably assisted by about a five times higher concentration of HPβCD compared to DNA used in the solution. Thus, from the experimental results, it is very evident that in the dye–HPβCD–DNA ternary system at pH 8.5, the dye can be preferentially bound to the HPβCD cavity in its neutral form Ac, using a reasonably high concentration of the macrocyclic host and thereby resisting the binding of the dye to the DNA host.
 | ||
| Fig. 9 (a) Normalized absorption spectra for the Ac–DNA–HPβCD, Ac–HPβCD, Ac and Ac–DNA systems at pH 8.5. [Ac] = 11 μM, [HPβCD] = 10 mM and DNA = 2.0 mM. (b) Normalized SS fluorescence spectra for the Ac–HPβCD, Ac–HPβCD–DNA, Ac and Ac–DNA systems at pH 8.5. [Ac] = 11 μM, [HPβCD] = 10 mM and DNA = 2.0 mM. | ||
 | ||
| Fig. 10 . (a) Normalized absorption spectra for the AcH+–DNA–HPβCD, AcH+–DNA, AcH+–HPβCD and AcH+ systems at pH 4. [AcH+] = 11 μM, [HPβCD] = 10 mM and DNA = 2.0 mM. (b) Normalized SS fluorescence spectra for the AcH+–DNA, AcH+–DNA–HPβCD, AcH+–HPβCD and AcH+ systems at pH 4. [AcH+] = 11 μM, [HPβCD] = 10 mM and DNA = 2.0 mM. | ||
As indicated from Fig. 10a and b, absorption and fluorescence spectral features of the dye–HPβCD–DNA ternary system at pH 4 match quite well with those of the dye–DNA system, signifying that the binding feature of the dye has now been tuned at this lower pH for preferential binding to the DNA pocket as the AcH+ form of the dye. From the present results, it is clearly evident that HPβCD can be applied as a useful pH-sensitive delivery nano-vehicle for the supply of the dye–drug to target DNA for a programmed acidolysis. The fact that the HPβCD macrocycle shows negligible or no interaction with either the free AcH+ or the AcH+–DNA complex is also evidenced from the results shown in the inset of Fig. 10b where there is hardly any observable difference in the fluorescence spectra between the AcH+ only and AcH+–HPβCD systems and also between the AcH+–DNA and AcH+–DNA–HPβCD systems.
 | ||
| Fig. 11 Circular dichroism spectra of the (a) Ac–HPβCD, (b) Ac–DNA and (c) Ac–HPβCD–DNA systems (pH 8.5). Concentrations of the components were: [Ac] = 50 μM, [HPβCD] = 20 mM and [DNA] = 1.2 mM. | ||
Circular dichroism spectra were also recorded in this study for the AcH+–HPβCD, AcH+–DNA and AcH+–HPβCD–DNA systems at pH 4 and the results are shown in Fig. S8 of the ESI.† As expected, the free AcH+ did not show any circular dichroism. For the AcH+–HPβCD system (cf. Fig. S8a, ESI†), there is also no observable ICD signal throughout the spectral region of the dye, suggesting no significant interaction of AcH+ with the HPβCD host. For the AcH+–DNA system, there is a positive ICD signal (cf. Fig. S8b, ESI†), albeit with much weaker intensity than that of the Ac–DNA system (cf.Fig. 11b), possibly suggesting that at pH 4 only a small fraction of the AcH+ undergoes intercalative binding and results in the observed weak ICD signal while the majority of the AcH+ undergoes electrostatic binding to the phosphate backbone of DNA and does not contribute to the ICD signal. For the AcH+–HPβCD–DNA ternary system, the ICD spectrum (cf. Fig. S8c, ESI†) is quite similar to that of the AcH+–DNA system (cf. Fig. S8b, ESI†), suggesting that at pH 4 the AcH+ form of the dye remains almost exclusively bound to DNA even in the presence of a significantly high concentration of the HPβCD host.
 | ||
| Fig. 12 (a) Fluorescence decay measured at 490 nm for the AcH+, AcH+–HPβCD, AcH+–HPβCD–DNA and AcH+–DNA systems at pH 4. [AcH+] = 11 μM, [HPβCD] = 10 mM and DNA = 2.0 mM. (b) Fluorescence decay measured at 410 nm for the Ac, Ac–DNA, Ac–HPβCD and Ac–HPβCD–DNA systems at pH 8.5. [Ac] = 11 μM, [HPβCD] = 10 mM and DNA = 2.0 mM. IRF represents the instrument response function. | ||
| System | pH | A 1 (%) | τ 1 (ns) | A 2 (%) | τ 2 (ns) | A 3 (%) | τ 3 (ns) |
|---|---|---|---|---|---|---|---|
| a The error limit in the lifetime values is about 5%. b These shorter lifetime components needed to be fixed for consistency with the proposed model and to obtain a good fit to the observed decays. | |||||||
| AcH+ | 4 | 100 | 32.0 | ||||
| AcH+–DNA | 4 | 96 | 29.0 | 4 | 1.52 | ||
| AcH+–HPβCD | 4 | 100 | 31.4 | ||||
| AcH+–HPβCD–DNA | 4 | 96.5 | 28.9 | 3.5 | 1.6 | ||
| Ac | 8.5 | 100 | 9.83 | ||||
| Ac–DNA | 8.5 | 79 | 8.65 | 21 | 0.48 | ||
| Ac–HPβCD | 8.5 | 46 | 8.95 | 54 | 2.27 | ||
| Ac–HPβCD–DNA | 8.5 | 34 | 8.6 | 52 | 2.27 (fixed)b | 14 | 0.48 (fixed)b |
For the dye–DNA system at pH 4, the decay is bi-exponential in nature (cf.Fig. 12a). In this case, the longer lifetime component (τ1 ∼ 29 ns) is marginally shorter than that of the free dye (∼32 ns) and is attributed to the electrostatic binding (exo- or semi-intercalative binding) of AcH+ to DNA. The shorter lifetime component (τ2 ∼ 1.52 ns), which is drastically shorter than that of the free dye and shows only a small contribution to the overall decay is understandably due to the fraction of the AcH+ intercalated between the DNA bases.33,54 The observation that the contribution of the longer lifetime component is very high (a1 ∼ 96%) in the present case suggests that at acidic pH the major fraction of the AcH+ undergoes exo- or semi-intercalative binding to DNA, due to the strong electrostatic interaction between the cationic dye and the negative phosphate groups of the DNA backbone. Such an inference is also supported by the results obtained from the circular dichroism studies, discussed in Section 1.3.2, and the TR fluorescence anisotropy studies, discussed in the next section. The reduction in the fluorescence lifetime compared to that of the free dye is regarded to be due to a quenching process caused by the nucleobases, possibly through a photoinduced electron transfer interaction,33,54–56 which is understandably very strong for the intercalated dye compared to the externally-bound dye.
Fluorescence decay for the dye–DNA system at pH 8.5 (cf.Fig. 12b) is also bi-exponential in nature, with a longer lifetime component (τ1 ∼ 8.65 ns) that is marginally shorter and a shorter lifetime component (τ2 ∼ 0.48 ns) that is drastically shorter than that of the free dye (∼9.83 ns). Since no appreciable electrostatic interaction is expected for the neutral Ac form of the dye with DNA, we anticipate that shorter and longer lifetime components at pH 8.5 correspond to the intercalated and the free dyes, respectively, for which supporting evidence is obtained from the fluorescence anisotropy results discussed in the next section. It should be mentioned here that for both AcH+ and Ac, the percentage reduction in the fluorescence lifetime due to DNA intercalation is very similar, about 94–95% compared to that of the free dye. Therefore, the observation that the shorter lifetime component has a greater contribution at pH 8.5 (21%) than at pH 4 (4%) suggests that the intercalation of the neutral Ac form of the dye into DNA is at least five times more favoured than that of the protonated AcH+ form of the dye.
For the dye–HPβCD system, the fluorescence decay also exhibits a bi-exponential nature at pH 8.5 (cf.Fig. 12b). The shorter lifetime component (τ2 ∼ 2.27 ns) is justifiably assigned to Ac encapsulated into the HPβCD cavity where a large reduction in the fluorescence lifetime compared to that of free Ac (∼9.83 ns) arises due to strong fluorescence quenching caused by the H-bonding interaction of the bound dye with the portal OH groups of the HPβCD host.19 The longer lifetime component (τ1 ∼ 8.95 ns), which is just marginally shorter than that of the solution with the dye alone (∼9.83 ns) is suggested to be due to the small fraction of the free dye that undergoes a dynamic fluorescence quenching by the HPβCD host, possibly involving a similar hydrogen bonding interaction. It should be mentioned that a similar quenching interaction for acridine dye has already been reported in our earlier study involving a parent βCD host.19
Interestingly, the fluorescence decay for the Ac–HPβCD–DNA ternary system at pH 8.5 resembles quite closely that of the Ac–HPβCD system but differs very greatly from that of the Ac–DNA system (cf.Fig. 12b). It is thus evident from the comparison of the fluorescence decays that in mildly basic solution, the Ac form of the dye in the Ac–HPβCD–DNA ternary system can be made to bind preferentially to the HPβCD host, even in the presence of a substantial concentration of DNA. In the present case, since multiple emissive species are present in the solution (cf.Table 1), the decay trace understandably required at least a tri-exponential function to fit acceptably and to be consistent with the proposed model, where the two shorter lifetime components correspond to the dye–DNA (τ2 = 0.48 ns) and dye–HPβCD complexes (τ3 = 2.27 ns) and the longer lifetime component (τ1 = 8.6 ns) represents the fraction of the free dye that undergoes dynamic quenching jointly by the HPβCD and DNA hosts present in the solution.
Fluorescence decays for the dye at different dye–host combinations at pH 4 are shown in Fig. 12a. As already mentioned, at pH 4 the fluorescence decay of the dye in the absence of any host is single-exponential in nature, with a lifetime of about 32 ns. For the dye–HPβCD system at pH 4, the decay interestingly shows a single-exponential nature, with only a small but quite systematic reduction in the lifetime with an increase in the HPβCD concentration. This observation suggests that the AcH+ form does not undergo any appreciable inclusion complex formation but the free excited dye undergoes dynamic quenching to a small extent by the HPβCD host. An important observation to be noted from Fig. 12a is that the decay for the AcH+–DNA–HPβCD ternary system exactly superimposes upon that of the AcH+–DNA system, confirming that at acidic pH the dye is completely bound to the DNA host as its AcH+ form. The observed TR fluorescence results are thus in complete correspondence with the results obtained from the ground state absorption and SS fluorescence studies, clearly indicating that in the dye–HPβCD–DNA ternary system the HPβCD in effect acts as a receptor to bind and stabilize the dye at higher pH and renders a complete relocation of the dye to the desired DNA target just upon reduction of the pH of the solution to the acidic region. In other words, HPβCD acts as an efficient nano-transporter to carry the dye–drug to the target DNA and delivers the model drug to the target upon using the drop in pH as the stimulus.
 | ||
| Fig. 13 (a) Time-resolved anisotropy decay curves for the AcH+, AcH+–HPβCD, AcH+–HPβCD–DNA and AcH+–DNA systems at pH 4. (b) Time-resolved anisotropy decay curves for the Ac, Ac–DNA, Ac–HPβCD and Ac–HPβCD–DNA systems at pH 8.5. | ||
| System | pH | r 1,0 | τ r1 (ps) | r 1,0 | τ r2 (ps) |
|---|---|---|---|---|---|
| a The error limit in the lifetime values is about 5%. | |||||
| AcH+ | 4 | 0.32 | 90 | ||
| AcH+–HPβCD | 4 | 0.32 | 91 | ||
| AcH+–DNA | 4 | 0.31 | 138 | ||
| AcH+–HPβCD–DNA | 4 | 0.32 | 133 | ||
| Ac | 8.5 | 0.31 | 89 | ||
| Ac–HPβCD | 8.5 | 0.17 | 95 | 0.14 | 760 |
| Ac–DNA | 8.5 | 0.28 | 89 | 0.03 | 910 |
| Ac–HPβCD–DNA | 8.5 | 0.18 | 96 | 0.14 | 770 |
As expected, the anisotropy decays for the free dye are very fast and follow mono-exponential kinetics both at pH 4 and 8.5, giving rotational time constants (τr1) of about ∼90 ps for both the AcH+ (pH 4) and Ac (pH 8.5) forms of the dye. At pH 4, the decay for the AcH+–HPβCD system also follows mono-exponential kinetics and gives a τr1 value (∼90 ps) very similar to that of the free AcH+. It is thus evident that there is no appreciable interaction of AcH+ with the HPβCD host, as also inferred from the SS and TR fluorescence and the circular dichroism results. Contrary to the AcH+–HPβCD system, for the AcH+–DNA system at pH 4, the anisotropy decay is slower and fits well with a mono-exponential function giving a τr1 value (∼140 ps) significantly higher than that of the free dye, although the increase is not exceedingly large. Recalling the TR fluorescence results in Table 1, it was indicated that in the AcH+–DNA system at pH 4 there is a longer lifetime component τ1 that shows the largest contribution (∼96%) and was attributed to the AcH+ exo-bound with DNA, while the shorter lifetime component τ2, showing only a small contribution (∼4%), was ascribed to the AcH+ intercalatively-bound to DNA. In accordance with these TR fluorescence results, it is expected that the anisotropy decay for the AcH+–DNA system at pH 4 is mainly due to the exo-bound AcH+ and hence should effectively follow a mono-exponential function.57 Since the AcH+ exo-bound with DNA is expected to suffer only a small retardation in its rotational motion, the τr1 value for the present system is accordingly increased only to a small extent compared to that of the free dye.
At pH 8.5, for both the Ac–HPβCD and Ac–DNA systems, the anisotropy decays follow bi-exponential kinetics. In both the cases, the τr1 component is very similar to that of the free dye while the τr2 component is drastically higher in comparison to τr1. As observed in the TR fluorescence studies (cf.Table 1), for the present systems the fluorescence decays are dominated by a longer lifetime component τ1 attributed to the fraction of the free dye present in the solution. The observation that the τr1 component for both the Ac–HPβCD and Ac–DNA systems resembles that of the free dye thus directly supports the proposition made from the TR fluorescence results. That the τr2 component for both the dye–HPβCD and dye–DNA systems is much higher than τr1 clearly suggests that this component is due to the Ac bound to the respective hosts, via inclusion complex formation with the HPβCD cavity and through intercalative binding to the DNA host. This is further supported by the fact that the τr2 value for the Ac–DNA system is much higher than that of the Ac–HPβCD system, understandably because the rotational motion of the dye intercalatively-bound to DNA will be much more restricted in comparison to that of the dye incorporated into the HPβCD cavity. Further, from the comparison of the τr2 value (910 ps) for the Ac–DNA system at pH 8.5 with the τr1 value (140 ps) for the AcH+–DNA system at pH 4 it is evident that the rotational motion of the DNA-bound Ac form is much more retarded than that of the DNA-bound AcH+ form of the dye. This is in direct support of our inference that in the AcH+–DNA system at pH 4, the cationic AcH+ form of the dye mainly undergoes an exo-binding to DNA through an electrostatic interaction whereas in the Ac–DNA system at pH 8.5 the neutral Ac form of the dye mainly undergoes an intercalative mode of binding with the DNA host. For the dye–HPβCD system at pH 8.5, the longer τr2 component is certainly due to the inclusion of Ac into the HPβCD cavity, which causes a large increase in the hydrodynamic volume of the fluorophore and hence an increase in the rotational time constant.
For the Ac–HPβCD–DNA ternary system at pH 8.5, the estimated anisotropy decay parameters (cf.Table 2 and Fig. 13b), match quite closely with those obtained for the Ac–HPβCD system. These results strongly support our proposition that in the Ac–HPβCD–DNA ternary system at pH 8.5 a major fraction of the Ac actually remained bound to the HPβCD cavity, even in the presence of a significantly high concentration of DNA. On the contrary, at acidic pH, the anisotropy decay for the AcH+–HPβCD–DNA ternary system interestingly resembles quite closely with that of the AcH+–DNA system (cf.Fig. 13a and Table 2). This observation directly reinstates the fact that at acidic pH the AcH+ form of the dye almost exclusively binds to the DNA host, even in the presence of a substantially high HPβCD concentration. Thus, the observed results in effect suggest that, depending upon the pH of the solution, the HPβCD host not only selectively binds but also releases the active dye to the target DNA site, induced by a suitable pH change.
In brief, the fluorescence anisotropy results correspond very nicely with the ground state absorption and the SS and TR fluorescence results and establish the fact that reduction in the pH of the solution can provide a simple stimulus to quantitatively release the dye from the HPβCD cavity for its binding to the target DNA in a dye–HPβCD–DNA ternary system. Such a strategy of controlled dye–drug release mechanism can be easily visualized by the schematic presentation shown in Scheme 3. From the present model study it is evident that the HPβCD nanocarrier can effectively be used for controlled and targeted release of potential anticancer prodrugs under acidic conditions and thereby to selectively increase the local drug concentration at the targeted site, reducing the untoward side effect of the drug, an advantage that is highly desired in chemotherapeutic applications.
 | ||
| Scheme 3 Schematic representation of the pH-induced transfer of acridine from HPβCD to DNA at acidic pH and vice versa. | ||
Conclusions
Interaction of an anticancer-active acridine dye with HPβCD, DNA and a HPβCD–DNA mixture has been investigated, using ground state absorption, steady-state fluorescence, time-resolved fluorescence, fluorescence anisotropy decay, 1H NMR and circular dichroism measurements at different pH conditions. Results show that HPβCD selectively and strongly encapsulates the neutral dye Ac but shows almost no affinity for the protonated dye AcH+. On the contrary, DNA interacts strongly with both the AcH+ and Ac forms of the dye, albeit with a somewhat higher affinity for the Ac form. Consequently, the differential binding of the two prototropic forms of the acridine dye observed with the HPβCD and DNA hosts leads to about 1.0 units of downwards pKa shift for the dye–HPβCD system and about 0.4 units of downwards pKa shift for the dye–DNA system. Using the acridine dye–HPβCD–DNA ternary system as a model, we demonstrate in this study the interesting strategy of pH-dependent formation and dissociation of the dye–HPβCD complex, for a targeted and controlled release of the dye from the nanocavity of the biocompatible model drug carrier HPβCD to the target DNA site. In the dye–HPβCD–DNA ternary supramolecular system, HPβCD not only selectively binds and stabilizes the neutral Ac form of the dye at mildly alkaline pH but also explicitly delivers the active protonated AcH+ form of the dye to the target DNA, triggered by a decrease in the pH to mildly acidic conditions. Thus, HPβCD acts as a model drug delivery agent to carry the dye to the target acceptor DNA. The potential of this unique supramolecular assembly may be explored in real biological systems where acidolysis can be used as a trigger for the controlled dissociation of the prodrug from the HPβCD inclusion complexes, leading to the association of the active form of the drug to the target site, such as to tumour cells or at the acidic organelles region, making the dye–drug more effective for the desired activity, with maximum selectivity. This model study would be very beneficial in relation to chemotherapeutic applications, nanoreactors, pharmaceutical development and can also be useful in designing new and promising stimuli-responsive assemblies for different biomedical applications. Future studies aim to investigate more into the interaction of anticancer acridine drugs with potential cyclodextrin derivatives for real pH-responsive biomedical applications.Experimental section
Acridine dye (purity >99%) was obtained from Sigma-Aldrich, USA, and purified by re-crystallization from cyclohexane solution. Hydroxypropyl-β-cyclodextrin (HPβCD, cf.Scheme 1; average degree of substitution = 0.8, average MW = 1500) was purchased from Aldrich and used as received. Calf thymus DNA (ct-DNA) was purchased from Sigma and used as received. Nanopure water (Barnstead System; conductivity of 0.1 μS cm−1) was used for all solution preparations. The dye solution was prepared by directly dissolving the dye sample and estimating its concentration from absorption spectral measurement (extinction coefficient 17793 M−1 cm−1 at λmax 355 nm for AcH+ and 9700 M−1 cm−1 at λmax 355 nm for Ac).19 A known weight of HPβCD was directly added to the experimental solution to achieve the required host concentration. Stock ct-DNA solution was prepared by dissolving the solid DNA sample in nanopure water and keeping the solution stored overnight at 4 °C. Each time fresh DNA solution was prepared to perform the experiments. As ct-DNA has quite a large distribution in molecular sizes (8.0–15 kb), the concentration of DNA in the experimental solution was determined in terms of total nucleotide concentration, following absorbance at 260 nm and using an extinction coefficient of 6600 M−1 cm−1.58–60 That the DNA sample is free from protein impurities was ensured by observing the ratio of absorbance at 260 nm to that at 280 nm to be in the range of 1.8–1.9.58–60
All the measurements were carried out in aqueous solutions at suitable pH conditions at ambient temperature (∼25 °C). The pH of the solutions was adjusted by adding dilute perchloric acid or dilute sodium hydroxide in small steps and the pH was measured using a pH meter (CL/46, Toshcon, India). Ground state absorption spectra and steady-state fluorescence spectra were recorded using a Jasco UV-visible spectrophotometer (model V-650; Tokyo, Japan) and a Hitachi spectrofluorimeter (model F-4500; Tokyo, Japan), respectively. Time-resolved fluorescence measurements were carried out using a time-correlated single-photon-counting (TCSPC) spectrometer,57,61 obtained from IBH, UK. In these measurements, a 374 nm diode laser (pulse width ∼100 ps, repetition rate 1 MHz) was used as the excitation source and a special photomultiplier tube (PMT)-based detection module supplied by Horiba Jobin Yvon IBH was used for the fluorescence detection. The instrument response function (IRF) for the TCSPC setup was measured using scattered light from a TiO2 suspension in water and the full width half maximum (FWHM) of the IRF was found to be ∼120 ps. All the measurements were carried out at a magic angle configuration to eliminate the effect of rotational anisotropy on the observed fluorescence decays. Observed decays were in general analyzed as a sum of exponentials (cf.eqn (2)), following a reconvolution procedure.57,61
| I(t) = ∑Biexp(−t/τi) | (2) |
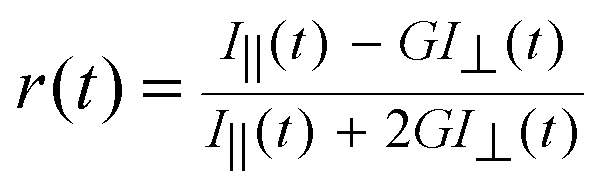 | (3) |
Acknowledgements
The authors are thankful to Dr A. Barik of RPCD and Dr S. Dey of Chemistry Division, BARC, for their help in the circular dichroism and NMR measurements, respectively. The authors also thankfully acknowledge the Bhabha Atomic Research Centre, Mumbai, India, for the generous support provided during the course of the present research work.Notes and references
- R. L. Carrier, L. A. Miller and I. Ahmed, J. Controlled Release, 2007, 123, 78–99 CrossRef CAS PubMed.
- K. Uekama, F. Hirayama and T. Irie, Chem. Rev., 1998, 98, 2045–2076 CrossRef CAS PubMed.
- D. W. Kuykendall and S. C. Zimmerman, Nat. Nanotechnol., 2007, 2, 201–202 CrossRef CAS PubMed.
- G. Astray, C. Gonzalez-Barreiro, J. C. Mejuto, R. Rial-Otero and J. Simal-Gandara, Food Hydrocolloids, 2009, 23, 1631–1640 CrossRef CAS PubMed.
- Z. Qi and C. A. Schalley, Acc. Chem. Res., 2014, 47, 2222–2233 CrossRef CAS PubMed.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS PubMed.
- E. M. M. D. Valle, Process Biochem., 2004, 39, 1033–1046 CrossRef.
- K. Langa, J. Mosinger and D. M. Wagnerová, Coord. Chem. Rev., 2004, 248, 321–350 CrossRef PubMed.
- H. Yamaguchi, T. Ogoshi and A. Harada, Sensor Development Using Existing Scaffolds, in Chemosensors: Principles, Strategies, and Applications, ed. B. Wang and E. V. Anslyn, John Wiley & Sons, New Jersey, 2011, ch. 11, pp. 211–226 Search PubMed.
- M. Shaikh, S. D. Choudhury, J. Mohanty, A. C. Bhasikuttan, W. M. Nau and H. Pal, Chem. – Eur. J., 2009, 15, 12362–12370 CrossRef CAS PubMed.
- M. Shaikh, J. Mohanty, A. C. Bhasikuttan, V. D. Uzunova, W. M. Nau and H. Pal, Chem. Commun., 2008, 3681–3683 RSC.
- G. Chen and M. Jiang, Chem. Soc. Rev., 2011, 40, 2254–2266 RSC.
- J. Zhang and P. X. Ma, Adv. Drug Delivery Rev., 2013, 65, 1215–1233 CrossRef CAS PubMed.
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed.
- Y. Cao, X. Hu, Y. Li, X. Zou, S. Xiong, C. Lin, Y. Shen and L. Wang, J. Am. Chem. Soc., 2014, 136, 10762–10769 CrossRef CAS PubMed.
- V. T. Perchyonok and T. Oberholzer, Curr. Org. Chem., 2012, 16, 2365–2378 CrossRef CAS.
- T. Loftsson and D. Duchene, Int. J. Pharm., 2007, 329, 1–11 CrossRef CAS PubMed.
- W. Al-Soufi, B. Reija, M. Novo, S. Felekyan, R. Kuhnemuth and C. A. M. Seidel, J. Am. Chem. Soc., 2005, 127, 8775–8784 CrossRef CAS PubMed.
- M. Shaikh, Y. M. Swamy and H. Pal, J. Photochem. Photobiol., A, 2013, 258, 41–50 CrossRef CAS PubMed.
- M. Shaikh, J. Mohanty, P. K. Singh, W. M. Nau and H. Pal, Photochem. Photobiol. Sci., 2008, 7, 408–414 CAS.
- V. Khorwal, B. Sadhu, A. Dey, M. Sundararajan and A. Datta, J. Phys. Chem. B, 2013, 117, 8603–8610 CrossRef CAS PubMed.
- S. Gould and R. C. Scott, Food Chem. Toxicol., 2005, 43, 1451–1459 CrossRef CAS PubMed.
- Z. Huang, S. Tian, X. Ge, J. Zhang, S. Li, M. Li, J. Cheng and H. Zheng, Carbohydr. Polym., 2014, 107, 241–246 CrossRef CAS PubMed.
- K. Miyake, H. Arima, F. Hirayama, M. Yamamoto, T. Horikawa, H. Sumiyoshi, S. Noda and K. Uekama, Pharm. Dev. Technol., 2000, 5, 399–407 CrossRef CAS PubMed.
- C. Liu, Z. Jiang, Y. Zhang, Z. Wang, X. Zhang, F. Feng and S. Wang, Langmuir, 2007, 23, 9140–9142 CrossRef CAS PubMed.
- A. Rescifina, C. Zagni, M. G. Varrica, V. Pistarà and A. Corsaro, Eur. J. Med. Chem., 2014, 74, 95–115 CrossRef CAS PubMed.
- S. Monti and I. Manet, Chem. Soc. Rev., 2014, 43, 4051–4067 RSC.
- X. Chen, L. Chen, X. Yao, Z. Zhang, C. He, J. Zhang and X. Chen, Chem. Commun., 2014, 50, 3789–3791 RSC.
- A. Rajendran and B. U. Nair, Biochim. Biophys. Acta, 2006, 1760, 1794–1801 CrossRef CAS PubMed.
- P. Belmont, J. Bosson, T. Godet and M. Tiano, Anti-Cancer Agents Med. Chem., 2007, 7, 139–169 CrossRef CAS.
- A. J. Pickard, F. Liu, T. F. Bartenstein, L. G. Haines, K. E. Levine, G. L. Kucera and U. Bierbach, Chem. – Eur. J., 2014, 20, 1–15 CrossRef PubMed.
- F. Charmantray, M. Demeunynck, D. Carrez, A. Croisy, A. Lansiaux, C. Bailly and P. Colson, J. Med. Chem., 2003, 46, 967–977 CrossRef CAS PubMed.
- K. Kawai, Y. Osakada, M. Fujitsuka and T. Majima, J. Phys. Chem. B, 2008, 112, 2144–2149 CrossRef CAS PubMed.
- H. W. Zimmermann, Angew. Chem., Int. Ed. Engl., 1986, 25, 115–130 CrossRef.
- R. W. Armstrongy, T. Kurucsev and U. P. Strauss, J. Am. Chem. Soc., 1970, 92, 3174–3180 CrossRef.
- M. K. Sarangi and S. Basu, Phys. Chem. Chem. Phys., 2011, 13, 16821–16830 RSC.
- V. Kumar, A. Pandey and S. Pandey, ChemPhysChem, 2013, 14, 3944–3952 CrossRef CAS PubMed.
- R. Wang, L. Yuan, H. Ihmels and D. H. Macartney, Chem. – Eur. J., 2007, 13, 6468–6473 CrossRef CAS PubMed.
- M. S. Ibrahim, I. S. Shehatta and A. A. Al-Nayeli, J. Pharm. Biomed. Anal., 2002, 28, 217–225 CrossRef CAS.
- G. Zhang, Y. Pang, S. Shuang, C. Dong, M. M. F. Choi and D. Liu, J. Photochem. Photobiol., A, 2005, 169, 153–158 CrossRef CAS PubMed.
- G. Zhang, X. Hu, N. Zhao, W. Li and L. He, Pestic. Biochem. Physiol., 2010, 98, 206–212 CrossRef CAS PubMed.
- C. Y. Huang, Methods Enzymol., 1982, 87, 509–525 CAS.
- M. Sayed, F. Biedermann, V. D. Uzunova, K. I. Assaf, A. C. Bhasikuttan, H. Pal, W. M. Nau and J. Mohanty, Chem. – Eur. J., 2015, 21, 691–696 CrossRef CAS PubMed.
- J. M. Schuette, T. T. Ndou, A. M. de la Pela, S. Mukundan Jr. and I. M. Warner, J. Am. Chem. Soc., 1993, 115, 292–298 CrossRef CAS.
- J. Carlstedt, A. González-Pérez, M. Alatorre-Meda, R. S. Dias and B. Lindman, Int. J. Biol. Macromol., 2010, 46, 153–158 CrossRef CAS PubMed.
- Y. Huang, Q. Lu, J. Zhang, Z. Zhang, Y. Zhang, S. Chen, K. Li, X. Tan, H. Lin and X. Yua, Bioorg. Med. Chem., 2008, 16, 1103–1110 CrossRef CAS PubMed.
- M. Ageno, E. Dore and C. Frontali, Biophys. J., 1969, 9, 1281–1311 CrossRef CAS.
- T. O'Connor, S. Mansy, M. Bina, D. R. McMillin, M. A. Bruck and R. S. Tobias, Biophys. Chem., 1981, 15, 53–64 CrossRef.
- C. M. Muntean, G. J. Puppels, J. Greve, G. M. J. Segers-Nolten and S. Cinta-Pinzaru, J. Raman Spectrosc., 2002, 33, 784–788 CrossRef CAS.
- F. Zsila, Z. Bikádi and M. Simonyi, Org. Biomol. Chem., 2004, 2, 2902–2910 CAS.
- F. Mendicuti and M. J. González-Álvarez, J. Chem. Educ., 2010, 87, 965–968 CrossRef CAS.
- M. Shaikh, J. Mohanty, M. Sundararajan, A. C. Bhasikuttan and H. Pal, J. Phys. Chem. B, 2012, 116, 12450–12459 CrossRef CAS PubMed.
- Q. Wang, R. He, X. Cheng and C. Lu, J. Inclusion Phenom. Macrocyclic Chem., 2011, 69, 231–243 CrossRef CAS.
- K. Fukui, K. Tanaka, M. Fujitsuka, A. Watanabe and O. Ito, J. Photochem. Photobiol., B, 1999, 50, 18–27 CrossRef CAS.
- K. Fukui and K. Tanaka, Angew. Chem., Int. Ed., 1998, 37, 158–161 CrossRef CAS.
- J. Joseph, N. V. Eldho and D. Ramaiah, Chem. – Eur. J., 2003, 9, 5926–5935 CrossRef CAS PubMed.
- J. R. Lakowicz, Principle of fluorescence spectroscopy, Plenum Press, Springer, New York, 2006 Search PubMed.
- H. Joshi, A. Sengupta, K. Gavvala and P. Hazra, RSC Adv., 2014, 4, 1015–1024 RSC.
- N. Nikolis, C. Methenitis and G. Pneumatikakis, J. Inorg. Biochem., 2003, 95, 177–193 CrossRef CAS.
- P. K. Singh and S. Nath, J. Phys. Chem. B, 2013, 117, 10370–10375 CrossRef CAS PubMed.
- D. V. O'Connor and D. Phillips, Time Correlated Single Photon Counting, Academic Press, New York, 1984 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c4cp05335d |
| This journal is © the Owner Societies 2015 |