
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHierarchical molecular dynamics of bovine serum albumin in concentrated aqueous solution below and above thermal denaturation†
Marco
Grimaldo
ab,
Felix
Roosen-Runge
a,
Marcus
Hennig
ab,
Fabio
Zanini
bc,
Fajun
Zhang
b,
Niina
Jalarvo
de,
Michaela
Zamponi
d,
Frank
Schreiber
b and
Tilo
Seydel
*a
aInstitut Max von Laue – Paul Langevin (ILL), CS 20156, F-38042 Grenoble, France. E-mail: seydel@ill.eu
bInstitut für Angewandte Physik, Universität Tübingen, D-72076 Tübingen, Germany
cMax-Planck Institute for Developmental Biology, Spemannstraße 35, D-72076 Tübingen, Germany
dJülich Centre for Neutron Science (JCNS), Forschungszentrum Jülich, D-52425 Jülich, Germany
eChemical and Engineering Materials Division, Neutron Sciences Directorate, and JCNS Outstation at the Spallation Neutron Source (SNS), Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 7th January 2015
Abstract
The dynamics of proteins in solution is a complex and hierarchical process, affected by the aqueous environment as well as temperature. We present a comprehensive study on nanosecond time and nanometer length scales below, at, and above the denaturation temperature Td. Our experimental data evidence dynamical processes in protein solutions on three distinct time scales. We suggest a consistent physical picture of hierarchical protein dynamics: (i) self-diffusion of the entire protein molecule is confirmed to agree with colloid theory for all temperatures where the protein is in its native conformational state. At higher temperatures T > Td, the self-diffusion is strongly obstructed by cross-linking or entanglement. (ii) The amplitude of backbone fluctuations grows with increasing T, and a transition in its dynamics is observed above Td. (iii) The number of mobile side-chains increases sharply at Td while their average dynamics exhibits only little variations. The combination of quasi-elastic neutron scattering and the presented analytical framework provides a detailed microscopic picture of the protein molecular dynamics in solution, thereby reflecting the changes of macroscopic properties such as cluster formation and gelation.
1 Introduction
Proteins are an essential part of the molecular basis of life. Their function relies on a combination of structure and dynamics.1–3 Therefore, a quantitative characterization of protein dynamics in their aqueous physiological environment on different length and time scales is an essential aspect for the understanding of the function of living systems. Most proteins possess a well-defined native globular structure, based on cooperative folding of the protein chain driven by an interplay of different intramolecular interactions of amino acid residues.4–7 Importantly, protein conformations display several structural features in a hierarchical order: starting from the sequence of amino acids—the primary structure—the protein chain builds structural elements such as α-helices, β-sheets or random coils—the secondary structure. These structural elements assemble into protein domains—the tertiary structure—that finally can arrange into multi-subunit proteins—the quaternary structure. As a consequence of the variety of typical time and length scales in such complex macromolecules, the dynamics of proteins is characterized by an extensive hierarchical nature.Upon increasing the temperature, thermal denaturation of the protein occurs. In this process, a partial unfolding of the native protein structure is followed by a reorganization to a different globular structure or a random-coil-state sometimes leading to aggregation and gelation.4–6,8–11 Regarding a single protein molecule, unfolding is related to protein malfunction,12 and a better understanding of the driving factors has important implications from a biomedical perspective. In protein solutions, denaturation represents a pathway how microscopic structural changes can alter macroscopic properties. In this context, gelation and formation of aggregates is of general importance for soft condensed matter, nanotechnology, and food research.
The first step of denaturation, the unfolding, is related to the general question of protein folding and misfolding and is not understood in detail, although reasonable predictions for small proteins exist and general features such as dominant contributions to folding and influences from the environment have been identified.7,12 In this context, the funnel picture of protein folding has gained significant importance in the past years and creates a need for dynamic measurements on the molten globule state.13,14 Mechanistically, Zimm and Bragg developed a theory for the first microscopic step of unfolding based on the helix-to-coil transition of a polypeptide chain as a result of which at low temperatures the polymer chain is dominated by helices, at high temperatures by random coils, with a rather narrow transition region where mixtures of these conformations may be found.4
While the Zimm–Bragg model takes only the secondary structure into account, real proteins possess a well-defined tertiary structure. Starting from a protein model with a dense core without contact to solvent and a hydrated outer shell, Finkelstein and Shakhnovich developed a theory of denaturation of single protein molecules and derived a first-order phase transition between the folded and a slightly expanded form.15,16 While this initial step of denaturation is independent of solvent, the further unfolding is dominated by the hydration-mediated interaction with the penetrating solvent,7 giving rise to a phase diagram for a single protein in dependence on solvent quality and temperature.15
Leaving the level of individual molecules, denaturation is represented by processes of aggregation and cross-linking, i.e. clustering and gelation, which, depending on the protein, can be rationalized either along the arguments of vulcanization of polymers17–19 or with colloidal gels with non-specific intermolecular attractive forces.20 A higher absolute concentration of the proteins has on the one hand a stabilizing effect on the protein thermal stability,21 on the other hand promotes the formation of intermolecular β-structures at lower temperatures.22
Numerous studies have focused on different aspects of the process of thermal denaturation by means of a variety of techniques.19,20,23–42 In the following, we focus on the effect of thermal denaturation on the internal dynamics of globular proteins.
Quasi-elastic neutron scattering (QENS) has been proven to be a well-suited tool to investigate the dynamics of proteins in aqueous solution on pico- to nanosecond time scales.43–47 Several elastic and quasi-elastic neutron scattering studies have compared the internal dynamics of proteins in native, molten (i.e. an intermediate equilibrium state between native and fully denatured), or denatured conformation, mostly in the picosecond time scale and mainly reflecting side-chain reorientations but in general not taking into account the global motion in solution.19,38,48–60 As a general outcome, the conformation-dependent dynamics is characterized by the increase of the number of mobile H-atoms on picosecond time scales and a decrease of the confinement of restricted motions in non-native structures. Importantly, upon unfolding, internal protein fluctuations exhibit increased dynamical heterogeneities.53,57 In this context, Gibrat et al. suggested that the dynamics of atoms within the protein is more influenced by the distance to the backbone than by solvent exposure.57
Importantly, thermal denaturation can lead to several steps of dynamical changes. Two characteristic temperatures for the denaturation of hydrated lysozyme were determined from the protein internal dynamics: above Tb = 325 K the mobility of side chains starts to increase, while Td = 346 K defines the limit for irreversible denaturation.41 Murayama and Tomida32 have shown that bovine serum albumin (BSA) in D2O at protein concentration cp = 20 mg ml−1 undergoes two conformational transitions at T = 330 K and around Td = 348 K. The latter corresponds to the melting of α-helices and the formation of intermolecular β-sheets. Above Td, the fraction of random coils, turns and connecting loops becomes at least as significant as that of α-helices and β-sheets.32 This finding, together with the fact that backbone fluctuations are much smaller in α-helices and β-sheets, compared to the greater amplitudes in connecting loops and unstructured termini,53 is consistent with the experimental observation that the average internal mean-square displacement of 500 mg ml−1 BSA in D2O increases faster for T ≳ 343 K.19
Reconsidering the hierarchical structure of proteins, a similar hierarchy in dynamics is expected, however, only partly suggested by the aforementioned studies on the average internal dynamics of entire proteins or labeled protein domains. In this article, we present a comprehensive study outlining the hierarchical protein dynamics before and after thermal denaturation. Using a high-quality data set recorded on the backscattering spectrometer BASIS61 (SNS, ORNL), we access the dynamics of BSA at nanosecond time and nanometer length scales in a temperature range between 280 and 370 K. We propose and justify a model reflecting the complex dynamics of the proteins. In terms of this model, we obtain three self-diffusion coefficients which we attribute to three hierarchical levels of the dynamics of proteins: (i) entire-protein translation and rotation, (ii) backbone fluctuations and (iii) side-chain motion. Consistently, the elastic incoherent structure factor (EISF) can be fitted by an extension of the model in ref. 47 accounting for the restricted dynamics of both side-chains and backbone. The analysis of the EISF, although more sensitive to the background treatment and to the fraction of coherent scattering, confirms an increase of mobile atoms and indicates a weaker restriction of the backbone with increasing temperature, while that of the side-chains is constant within the error. Both our new model and its physical interpretation may not be complete or unique, but we find that previous models of protein diffusive dynamics are not consistent with our observations at high temperatures.
In the following, subsequent to the Experimental section, we present our results as well as a new analytical framework. This framework provides a quantitative access to the internal motions of proteins during the changeover from freely diffusing molecules in solution at low temperatures to protein molecules incorporated in a gel at high temperatures. Following the Results section, we discuss our observations in terms of the aforementioned hierarchical levels and in the context of the reported previous findings. In the Conclusions section, we draw a comprehensive picture of the nanosecond self-dynamics observed on molecular length scales.
2 Experimental section
Bovine serum albumin (BSA) was obtained from Sigma-Aldrich (product code A3059) as a lyophilized powder (99% purity) and used without further purification. The proteins were dissolved in water (D2O) and filled in double-walled cylindrical aluminum sample holders (outer diameter 22 mm, gap 0.25 mm) for the neutron scattering experiments. The cylinders where sealed against vacuum and inserted into a cryostat on the neutron spectrometer for temperature control.The neutron data were recorded on the backscattering spectrometer BASIS61 (SNS, ORNL). Neutrons were analyzed using Si(111) single crystals, thus setting the analyzed energy to 2.08 meV. The integration time for each neutron spectrum amounted to approximately 4 hours per temperature. We have carried out the data reduction using the Mantid software provided by the SNS and subsequently analyzed the data using MATLAB, as reported in detail in ref. 62.
Using neutron spectroscopy, we measure the scattering function S(q,ω) at scattering vectors q which are coupled to reciprocal intermolecular distances and at energy transfers ω which can be resolved, at BASIS, down to ∼3.5 μeV FWHM. We have determined the spectrometer resolution function ![[scr R, script letter R]](https://www.rsc.org/images/entities/char_e531.gif) by recording the scattering signal from a vanadium standard and modeled it by a sum of 4 Gaussians. We have subsequently implemented the convolution of with the fit functions in MATLAB analytically by building Voigt functions.62
by recording the scattering signal from a vanadium standard and modeled it by a sum of 4 Gaussians. We have subsequently implemented the convolution of with the fit functions in MATLAB analytically by building Voigt functions.62
3 Results and discussion
3.1 Model-free observations
Fig. 1 depicts the evolution of the measured scattering function S(q,ω) in dependence of temperature. In general, two temperature regimes can be observed directly from visual inspection. Below Td ≈ 345 K, the overall broadening of the spectra increases with rising temperature, indicating increasingly faster dynamics. Around Td, the spectral width decreases sharply. Finally, for T > Td, the spectral width appears to remain constant or increase slightly with increasing temperature. The elastic scattering intensity (ω = 0) decreases with rising T for T < Td and increases again for T > Td, consistent with earlier observations focusing on the elastic signal only.19 These model-free observations are consistent with the intuitive picture of denaturing. Before denaturing, the proteins diffuse freely in solution and global and internal dynamics become faster due to elevated temperatures. Upon denaturation, in particular the global dynamics irreversibly arrests due to unfolding and cross-linking between proteins, and the remaining dynamics again increases with rising temperature. This physical picture will be discussed further in the following subsections based on model fits that provide a consistent description of the observed dynamics.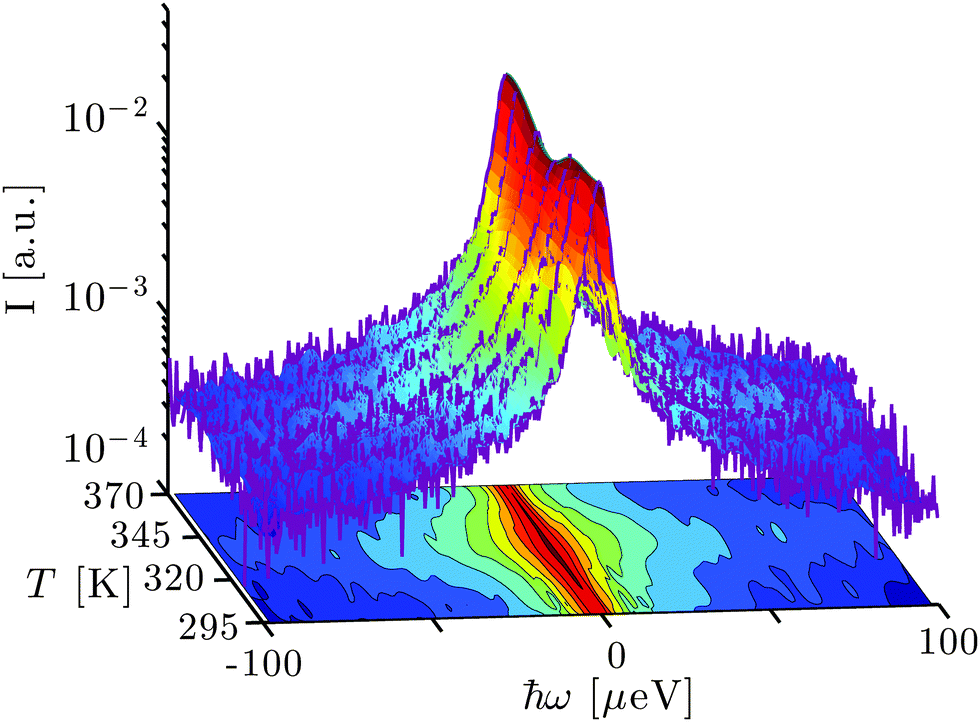 | ||
| Fig. 1 Example spectra (purple lines) recorded using the backscattering spectrometer BASIS, SNS, ORNL, on bovine serum albumin (BSA) at the scattering vector q = 0.85 Å−1 for different temperatures T at the protein concentration cp = 200 mg ml−1 in heavy water (D2O) subsequent to the subtraction of the empty can spectra. The surface interconnecting the 7 spectra is a spline interpolation of binned data (7 points per bin for |ω| > 10 μeV). The bottom projection displays the normalized scattering intensity S(q,ω)/S(q,ω = 0) using spline interpolation and illustrates the T-dependent width of the spectra. | ||
3.2 Analysis via the conventional two-Lorentzian model below the denaturation temperature
The short-time self-diffusion of proteins in concentrated solutions and its dependence on crowding has been investigated earlier at temperatures well below denaturation.46,63 Advances in the performance of neutron backscattering spectrometers have made it possible only very recently to address both the global protein self-diffusion and its internal dynamics from solution samples, and to systematically investigate the effect of crowding on these two types of dynamics.47 In that temperature range, the quasi-elastic scattering function has been shown to be well modeled byS(q,ω) = ⊗ {β(q)[A0(q)![[script L]](https://www.rsc.org/images/entities/char_e144.gif) γ(ω) + (1 − A0(q))γ+Γ(ω)] + βD2OγD2O(ω)} γ(ω) + (1 − A0(q))γ+Γ(ω)] + βD2OγD2O(ω)} | (1) |
denotes the instrumental resolution function, modeled by a combination of Gaussian functions, β(q) is a scalar, and A0(q) represents the elastic incoherent structure factor (EISF). The two Lorentzians γ(ω) and γ+Γ(ω) account for two processes occurring at distinct time scales, where the faster process, responsible for a broadening of the elastic line of width Γ, is convoluted to the slower process with line broadening γ. Finally, the fixed term βD2OγD2O models the solvent contribution. This fixed term is determined using time-of-flight spectroscopy data on pure D2O for the linewidth γD2O = γD2O(T) in combination with BASIS data on pure D2O for the amplitude βD2O = βD2O(T) as explained in detail in ref. 62.
We have carried out the full analysis both with and without the assumption of an H/D-exchange of the labile H-atoms on the proteins with the solvent water. We estimate the magnitude of this exchange based on the number 776 of labile H-atoms64 per BSA molecule obtained from the protein data base (PDB) file.65 Taking into account the molar fraction of BSA in water and the scattering lengths of H and D, respectively, we obtain an increase of the amplitude of the solvent water signal compared to the assumption of an absent H/D-exchange by a factor of 1.08, 1.16, and 1.38, respectively, for the protein concentrations of 100, 200, and 500 mg ml−1, respectively (resulting in 1, 2, and 5% H/D exchange in the solvent). Due to a remaining uncertainty in the exact number of exchangeable atoms, we display the figures without assuming the H/D-exchange in the article and the corresponding figures assuming the estimated exchange in the ESI.† The increase of the solvent scattering signal due to the H/D-exchange does not change the overall trend of the fit results, and we will report the essential fit results both with and without assumption of the exchange. In principle, the H/D-exchange does not only affect the solvent scattering amplitude, but also its dynamics. However, since the H/D-exchange amounts to only approximately 5% of the total number of hydrogens in the water for the case of the highest protein concentration, we assume that the effect of the modified solvent dynamics is small. The short-time dynamics of D2O is essentially unaffected by the formation of a gel,66 therefore the treatment of its contribution is valid both in the solution and in the gel states of our samples. Having carefully handled the contribution of D2O, we estimate that the incoherent fraction of the total scattering intensity is ∼88%, i.e. that of a dry protein powder, the coherent intensity varying only slightly in q.67 In the following, we will thus carry out the analysis neglecting the coherent contribution, being aware that this might introduce a small systematic error, in particular in the EISF.
An example spectrum (symbols) fitted with eqn (1) (solid line superimposed on the data) is shown in Fig. 2(a). The two Lorentzians γ and Γ are depicted by the blue and the green dashed lines respectively, and βD2OγD2O is represented by the magenta dotted line. In the physical picture established earlier,47 γ is associated with the global protein motions, and Γ is attributed to the internal molecular diffusive motions of the proteins. We find that the fitted γ are consistent with a Fickian process for the global self-diffusion, i.e. γ = dq2 with the apparent diffusion coefficient d (cf.Fig. 2(b)). Here, d is denoted ‘apparent’, because it contains contributions from both rotational and translational diffusion. The internal dynamics associated with the width Γ can be described by jump-diffusion for T < 340 K, i.e. Γ = Dq2/(1 + τDq2) with a diffusion coefficient D and residence time τ.68
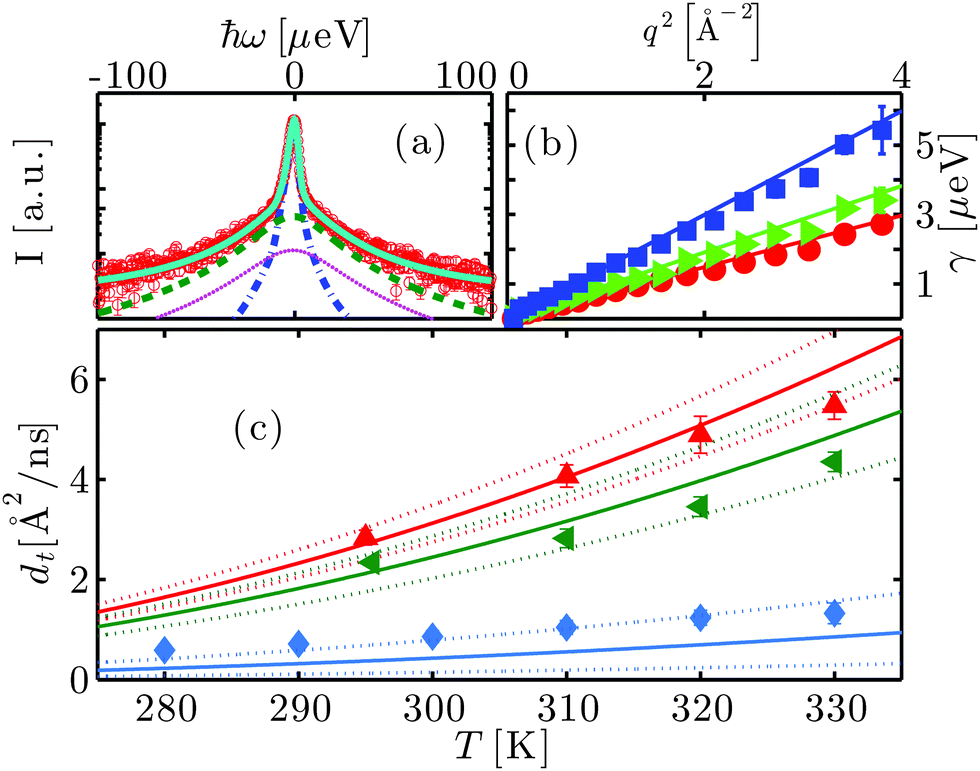 | ||
| Fig. 2 (a) Example spectrum (symbols) recorded using the neutron backscattering spectrometer BASIS, SNS, ORNL, on bovine serum albumin (BSA) at the concentration cp = 200 mg ml−1 in heavy water (D2O) at the temperature T = 295 K. The displayed data have been detected at the scattering vector q = 0.45 Å−1. The solid line superimposed on the data is a fit of the model function (eqn (1)) containing two Lorentzians accounting for the dynamics of the protein molecules (blue and green dashed lines) and a fixed contribution accounting for D2O (magenta dotted line), convoluted with the resolution function. (b) Linewidth γ of the Lorentzian accounting for the translational and rotational diffusion of the entire protein as a function of q2 for cp = 500 mg ml−1 at T = 280, 290 and 310 K (red circles, green triangles and blue squares, respectively). The lines are the fits following the Fickian law γ = dq2. (c) Translational self-diffusion coefficients dt as a function of temperature for cp = 150 mg ml−1 (red pointing-up triangles), 200 mg ml−1 (green triangles), and 500 mg ml−1 (blue diamonds). The solid lines are the respective theoretical self-diffusion coefficients and the dotted lines indicate the confidence interval defined by a relative error of 5% on the ratio between hydrodynamic radius RH and effective radius R. | ||
3.3 Analysis via a model of switching diffusive states at elevated temperatures
At higher temperatures, the fit results for Γ are no longer consistent with a jump-diffusion process such as that by Singwi and Sjölander68 or that by Hall and Ross:69 a marked nonmonotonicity of the q-dependent Γ resulting for a single process (see the ESI†) cannot be interpreted in physical terms in a straight-forward way. By contrast, a multi-state process would be consistent with such observation. Therefore, in order to extend the model given by eqn (1), we substituteΓ(ω) with the scattering function for a particle switching between two diffusive processes having rates Γ1 = D1q2 and Γ2 = D2q2 and residence times τ1 and τ2, respectively. D1 and D2 are thus the self-diffusion coefficients of the two states. Essentially, such a picture differs from that leading to the Singwi–Sjölander jump-diffusion model in that the (overdamped) particle vibration is substituted by a second diffusive state, and both diffusive states have a finite residence time. We have tested several other models as well, but these led to inconsistent results, as discussed in the ESI.† The scattering function for the two-state model reads:70| Ssw(q,ω) = αλ1(ω) + (1 − α) λ2(ω) | (2) |
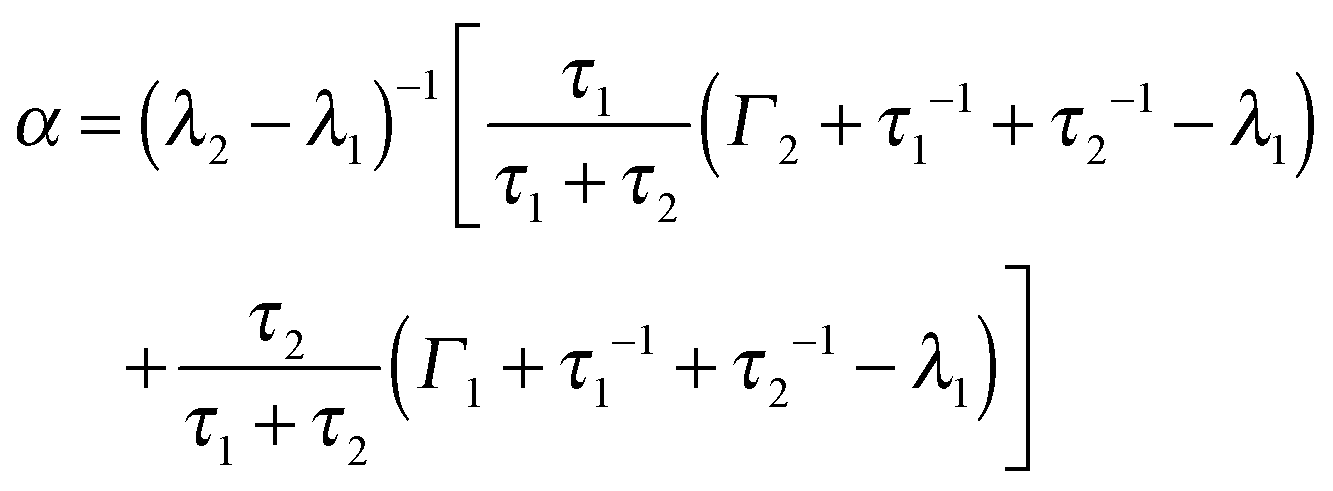 | (3) |
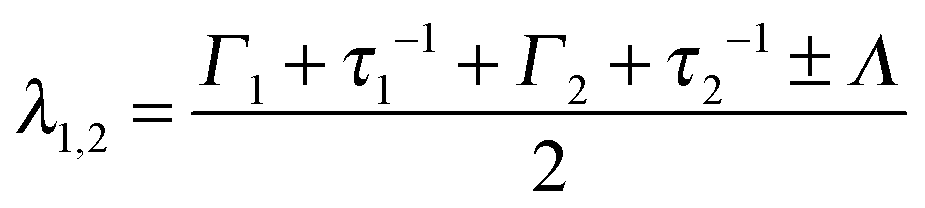 | (4) |
| Λ = [(Γ1 − Γ2 + τ1−1 − τ2−1)2 + 4(τ1τ2)−1] | (5) |
Thus, the overall scattering function in this picture is:
| S(q,ω) = ⊗ {β(q)[A0(q)γ(ω) + (1 − A0(q))(αγ+λ1(ω) + (1 − α)γ+λ2(ω))] + βD2OγD2O(ω)} | (6) |
Given the large number of free parameters, we set a priori Γ1,2 = D1,2q2 and γ = dq2, where d represents the apparent self-diffusion coefficient of the entire protein. We are thus assuming that, at the short time and length scales accessible by the instrument, the dynamics of the entire protein corresponds to simple self-diffusion even at higher temperatures, having reliably confirmed this for physiological T.46,47 We can then fit the model to the spectra in the entire q-range simultaneously with d, D1, D2, τ1 and τ2 as global parameters, while A0(q) and β(q) are allowed to change at every q. The fixed term βD2OγD2O models the solvent contribution,47,62 as explained in Section 3.2. An example spectrum fitted with eqn (6) as well as the global fit parameters as a function of T are plotted in Fig. 3. The fits show a very good agreement of data and switching model for the full temperature range. Furthermore, the fit parameters show small error bars, evidencing a reasonable statistical representation of the data by the model. In this context it is important to emphasize that, due to the global fit parameters, the switching model has fewer free parameters than the two-Lorentzian fit function on the full q range.
 | ||
| Fig. 3 (a) Apparent self-diffusion coefficient d as a function of T for cp = 150, 200 and 500 mg ml−1 (symbols) and fits with eqn (7) (lines). The illustrations depict a colloidal suspension of native proteins (left) and a cross-linked network of denatured proteins (right). (b) Example spectrum (symbols) of aqueous BSA at cp = 500 mg ml−1, T = 370 K and q = 0.45 Å−1. The solid line superimposed on the data is a fit of the model function (eqn (6)). The blue dot-dashed line depicts the Lorentzian describing the protein global self-diffusion, while the light green asterisks and the dark green dashed line represent the Lorentzians attributed to backbone and side-chain dynamics, respectively. The magenta dots denote the contribution of D2O. (c) D1 as a function of T (symbols). D1 is attributed to the dynamics of the backbone and sensitive to the secondary structure, part of which undergoes a transition from α-helix (left image) to disordered coil (right image). The illustration of a helix-to-coil transition was rendered with VMD.71 The lines are guides to the eye. (d) Arrhenius plot of the residence time τ1 between two jumps of the side-chains versus T for three cp (symbols). The data above denaturation were fitted with an Arrhenius equation (line), while at low temperatures the line is a guide to the eye. (e) D2 as a function of T for the concentrations reported in the legend (symbols). D2 is associated with the dynamics of the side-chains. The lines are guides to the eye. The illustrations depict solvent-inaccessible side-chains in the folded protein (left) becoming solvent-exposed in the unfolded protein (right). (f) Arrhenius plot of the residence time τ2 as a function of T (symbols). τ2 is the time a side-chain needs to switch between two conformations during which they are considered immobile with respect to the backbone. The data above denaturation were fitted with an Arrhenius equation (line). The red open triangles in (a), (c)–(f) refer to the sample at 500 mg ml−1 cooled back to room temperature after denaturation. | ||
In the following Sections we report the fit results, which we subsequently interpret in the context of a physical picture that has already been suggested in the model-free results. Generalizing the picture established for physiological temperatures,47 we will associate d with the global protein diffusion and the parameters D1, D2, τ1 and τ2 with internal dynamics.
3.4 Global apparent short-time self-diffusion
Fig. 3(a) shows the apparent self-diffusion coefficient d of the proteins at cp = 150, 200 and 500 mg ml−1 as a function of T (symbols). As expected, a higher cp causes a lower d due to the effect of crowding on the hydrodynamic interactions.46 This difference aside, the three concentrations exhibit analogous behaviors, consistent with elastic neutron scattering measurements:19d increases for T ≲ 340 K, decreases up to T ≈ 360 K and then increases again, with a smaller slope. As in ref. 19, we fit the data with the function| d(T) = (m1T + b1)[1 − Θ(T*)] + (m2T + b2)Θ(T*), | (7) |
3.5 Translational short-time self-diffusion below the transition temperature Td
Below the transition temperature Td, d can be used to calculate the translational self-diffusion coefficient dt once the structure of the protein is known.46 As already observed both for BSA and gammaglobulins,46,47dt(φ,T) can be well described by the theory of colloidal hard spheres72 (see Fig. 2(c)). For cp = 500 mg ml−1, dt is systematically higher than the curve of colloids. This is probably because at such high concentrations the determination of the effective protein volume fraction is less accurate. Also, at temperatures close to Td, dt becomes systematically lower than the theory for cp ≤ 200 mg ml−1. The effect, although significant, is rather small and can be probably attributed to thermal expansion that is difficult to quantify. Note that such a deviation can be caused by an increase of the hydrodynamic radius RH by only 3%, since the translational and rotational diffusion coefficients depend differently on RH46 (see also the ESI†).3.6 Internal dynamics
In addition to the global motion of the entire protein molecule, the wide energy range accessible at BASIS allows reliable fits of the parameters D1,2 and τ1,2 in eqn (6), which correspond to broader contributions in the spectra and in the physical picture are associated with internal motions. D1 is plotted in Fig. 3(c) as a function of T for the three investigated samples. Like d, also D1 seems to decrease monotonically with protein concentration, probably due to a crowding-induced stabilization effect.For all samples D1 remains nearly constant or only slightly increases for T < Td and grows faster for higher temperatures resembling the high-temperature dynamical transition observed in other studies.19,38 The residence time τ1 associated with this first internal diffusive state is, within the error bars, crowding-independent and is essentially constant at ∼100 ps for T < Td (Fig. 3(d)). At T ≈ Td, τ1 sharply increases by an order of magnitude to ∼1 ns. The further decrease of τ1 for T > Td can be modeled by an Arrhenius behavior with activation energy Ea ∼ 12 kcal mol−1 ≃ 0.5 eV (dashed line). (When assuming the increased amplitude of H-contaminated solvent water (Section 3.2), Ea ∼ 18 kcal mol−1 ≃ 0.8 eV, see ESI.†) Note that the sample at cp = 500 mg ml−1 has been measured at 300 K both before and after denaturation. Although d at 300 K is, as expected, much lower in the denatured sample, D1 is comparable before and after denaturation and τ1 is only slightly higher in the gel (open triangle), indicating that, on the accessible time and length scales, the associated diffusive state is not strongly affected by the macroscopic structure.
D 2 linearly increases with T up to Td, decreases stepwise for cp = 200 and 500 mg ml−1 and then increases again with a similar slope (cf.Fig. 3(e)). D2 is crowding-dependent, being smaller for higher protein concentrations. The residence time τ2 exhibits a behavior very similar to that of τ1, with τ2>τ1, and is independent of cp within the error bars (Fig. 3(f)). The activation energy calculated from τ2 for T > Td is Ea ∼ 13 kcal mol−1 ≃ 0.6 eV (Ea ∼ 12 kcal mol−1 ≃ 0.5 eV assuming the H-contaminated solvent, see above). Similarly to D1 and τ1, the values of D2 and τ2 for cp = 500 mg ml−1 after denaturation at T = 300 K are only slightly shifted to slower dynamics. In particular, D2 is somewhat lower and τ2 higher in the denatured sample, indicating a minute slowdown of this diffusive component. Thus, we obtain that the change of the overall internal dynamics on the investigated length and time scales varies only slightly for different folded protein structures, and more significantly for the unfolded structure, while being independent of the entire-protein dynamics. This result supports the validity of the decoupling of the global from the internal dynamics.
We note that we observe dynamics that is significantly faster than the surrounding water, as becomes apparent when comparing the fit results for D2 with the published values for the diffusion coefficient of water.62,73 Dynamics on similar or even faster time scales has been observed in related systems and attributed to side chain dynamics.74,75 The fast time scale can be rationalized in this picture by the fact that the side chain motions partly consist of fast rotations or reorientations of the chain, or of smaller subgroups such as methyl groups.
3.7 Geometry of confined motions
The EISF A0(q) in eqn (6) contains information on the geometry of localized motions within the protein. Fig. 4(a) and (b) depicts the EISFs as a function of q for the temperatures given in the legend and cp = 200 and 500 mg ml−1, respectively (symbols). At the lowest temperatures the EISF can be fitted well by the model proposed in a previous study on γ-globulins, i.e. a sum of two contributions from methyl-group rotations and diffusion in a sphere.47 By contrast, this model does not adequately describe our data when approaching and overcoming Td. Extending the aforementioned model, we fit the EISF with the function| A0(q) = p + (1 − p)AG(q,a)[sAsph(q,R) + (1 − s)A3−j(q,aM)] | (8) |
Here, s is the fraction of hydrogen atoms diffusing in an impermeable sphere, and (1 − s) is the fraction of hydrogens undergoing three-sites jump-diffusion attributed in the physical picture to methyl-group reorientations. p represents the fraction of H-atoms appearing fixed on the accessible time scale. Consequently, (1 − p) is the fraction of H-atoms moving on the accessible time scale. The three-sites jump diffusion is modeled by the term76,77
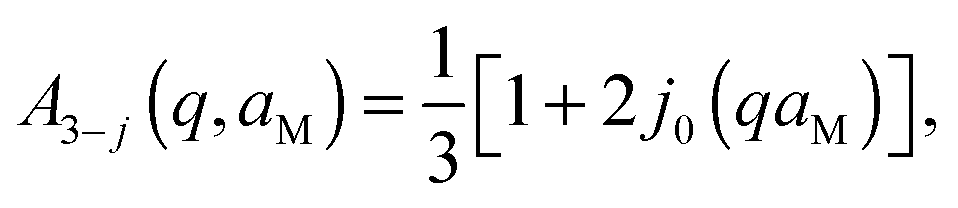 | (9) |
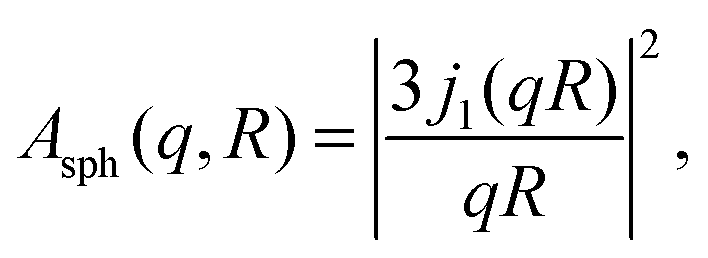 | (10) |
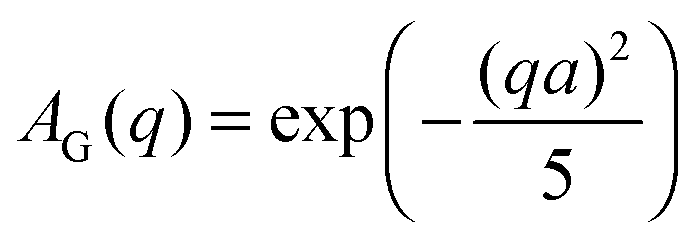 | (11) |
 | ||
| Fig. 4 (a) and (b): EISF as a function of q at the temperatures given in the legend for cp = 200 and 500 mg ml−1, respectively, (symbols) and fits following eqn (8) (solid lines). (c) Radius a (eqn (11)) as a function of T for cp = 500 mg ml−1 (symbols) and fit with eqn (12) (blue solid line). a is associated with the effective sphere accessible by backbone atoms. (d) Radius R (eqn (10)) as a function of T (symbols). The lines are guides to the eye. This radius defines the sphere accessible by side-chain motions. (e) Fraction of immobile atoms p as a function of T (symbols). The lines are guides to the eye. (f) s as a function of T (symbols) defining the ratio of side-chains describable with a diffusion in a sphere model to the total amount of mobile side-chains. The line is a guide to the eye. The parameters in Fig. 4(c)–(f) are obtained from the fit of the EISF with eqn (8), and the open symbols in Fig. 4(b)–(f) refer to the sample at cp = 500 mg ml−1 cooled down to room temperature after irreversible denaturation. | ||
The fits with eqn (8) are depicted by the lines in Fig. 4(a) and (b). The open triangles measured in the gel-like sample at 300 K deviate slightly from those measured in solution before denaturation (full blue triangles) only in the lower q-range, reflecting a decrease of the confinement only for the largest accessible length scales in the denatured proteins. For cp = 500 mg ml−1, the radius a of the effective sphere accessible by the backbone is shown in Fig. 4(c). If one assumes that backbone atoms sit in an elastic potential energy landscape, a can be fitted by
 | (12) |
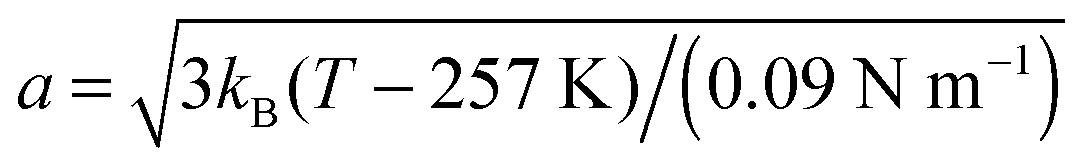 in eqn (8), thus reducing the number of free parameters to 3. Doing so, we assume that the geometry of the confined backbone fluctuations is essentially crowding-independent, although its dynamics varies with cp as suggested by our previous study on γ-globulins.47
in eqn (8), thus reducing the number of free parameters to 3. Doing so, we assume that the geometry of the confined backbone fluctuations is essentially crowding-independent, although its dynamics varies with cp as suggested by our previous study on γ-globulins.47
The parameters R, s and p obtained from the fit of the EISF are plotted in Fig. 4(d)–(f). The lines are guides to the eye. The radii R are roughly constant within the error bars, while as a general trend p decreases with temperature confirming earlier results.48–51,53–55,57,58 Also the trend of s is similar for all protein concentrations: s remains essentially constant for T < Td, reaches a sharp maximum about Td and then decreases again, possibly reaching a plateau. All parameters indicate that, once the sample is cooled down back to 300 K (open triangles) only minor discrepancies with the system before denaturation have occurred.
4 Discussion of the general physical picture
While summarizing the results presented above, we discuss in the following a possible interpretation as an overall picture for the hierarchical protein dynamics measured here. Before going into detail, we note that our data are unequivocally characterized by a dynamical transition around Td ≈ 342 K, consistent with the literature,19,42 probably coupled to the dynamical transition of hydration water.38,41,82 Another slightly lower characteristic temperature around Tb ∼ 330 K is visible in the behavior of p (Fig. 4(e)), above which structural changes are reported42 and side-chains increase their mobility.41We assign the three diffusive processes in the model given by eqn (6) (i.e. the processes associated with the diffusion coefficients d, D1, and D2, respectively) to three hierarchically distinct dynamics levels. The first of these is described by the short-time self-diffusion coefficient d and represents the dynamics of the entire protein. This process, although occurring at small time and length scales, is strictly related to macroscopic properties of the sample. It can be well described by the theory of colloids as long as the sample is still a solution, and becomes much slower when the gel is formed. When cooled down to room temperature after complete denaturation, the gel-like state is reflected in a much lower d than in the solution before heating up, indicating that the self-diffusion of entire proteins is strongly hindered e.g. through cross-linking or cluster formation.
The two-state model (eqn (2)) describes a dynamical system that switches between two states with diffusion coefficients D1,2 with constant switching rates 1/τ1,2. Given the different magnitudes of D1 and D2 and comparable magnitudes of τ1 and τ2, we speculate that the two diffusive states correspond to two scenarios of side-chain confinement with respect to the protein backbone. (i) Side-chains are blocked in a certain position relative to the backbone atoms of the respective amino-acid (e.g. due to steric hindrance of close-by side-chains or fluctuating backbone segments). In this case the side-chains move together with the backbone with the diffusion coefficient D1. (ii) The atoms of the side-chains are moving in a confined space limited mainly by the covalent bonds of the side-chain itself. These movements associated with the diffusion coefficient D2 are one order of magnitude faster than the backbone motion that can be therefore neglected here. Mobile side-chains switch between the two states with residence times τ1 and τ2. We note that we cannot rule out a small contribution from interfacial water, which might occur at a similar time scale as the side chain motions. We neglect this possible effect due to the low scattering signal of the deuterated solvent, and attribute the two dynamical processes to backbone and side-chain diffusion.
This interpretation is consistent with the model for the EISF, where the geometry of confinement of the side-chains is “smeared-out” by that of the backbone on the order of a ∼ 1 Å. Therefore, in this picture, we account for the heterogeneity of protein internal dynamics by separating slower and smaller-amplitude backbone fluctuations from faster and greater-amplitude side-chain motions, coherently with previous findings.28,29,36,53,57
The effect of temperature on the overall dynamics of proteins in solution at the observable time scales can be explained at different hierarchical levels as follows. For T < Td the proteins follow a center-of-mass diffusion in agreement with the theory of colloids. While the side-chain diffusivity increases linearly with T in this range, the effect on the dynamics of backbone fluctuations is rather limited. The effective radius a of the volume accessible to the backbone increases as a square-root, consistent with a spring model. The confinement radius R for the side chains remains constant within the errorbars, consistent with the length of a covalently bound side chain that determines the explored volume, rather than little fluctuations of atoms about its axis.
Around Td, significant changes of the dynamics at all three levels are observed. The entire protein starts to slow down, probably because of cluster formation and cross-linking between proteins. At the same time, the backbone undergoes a dynamic crossover, similar to that reported for hydration water and for the backbone root mean-squared displacement (RMSD) of hydrated powders of lysozyme in NMR experiments82 and simulations,38 and in the RMSD of BSA solutions from fixed window elastic neutron scattering.19 Such an observation can be attributed to a Zimm–Bragg type structural transition of the α-helices to random coils.4 The side-chain diffusivity follows a crowding-dependent behavior with a step-like reduction within a temperature range of ∼10 K for cp = 200 and 500 mg ml−1, not discernible at the lowest concentration. This may be related to the increase of the exposed side-chains upon unfolding and a higher damping in the denser gel. The increase of τ1 and τ2 by an order of magnitude might be related to the higher solvent-accessible surface as well: the presence of water molecules might on the one hand prolong the average time needed for a side-chain getting solvent-exposed to “flip” between equilibrium positions, and on the other hand prevent it from moving for a longer time while waiting that D2O density fluctuations allow it to escape the equilibrium position.
The geometry of confinement of both backbone and side-chains does not exhibit an equally pronounced transition, which is reasonable since the local confinement is mainly governed by the unaltered primary structure. The number of immobile atoms p decreases. The sharp increase of the fraction s of side-chains exploring an effective sphere with average radius R suggests that steric hindrance from other side chains decays due to changes of the secondary structure. We note that both fractions of mobile side-chains may include methyl-groups, but since R ≫ aM, their contribution can be neglected in a first approximation for the fraction s.
Further increasing T, proteins form bigger aggregates or become increasingly cross-linked into a gel-like state causing their global self-diffusion to decrease to a minimum. Proteins are now cross-linked through intermolecular β-sheets32 and the global short-time self-diffusion is defined by the motion of the segments between two cross-links. At a certain point, either the clusters are too strongly charged to grow further or the cross-link number saturates, and protein global motion starts to increase again with T. The decrease of s is consistent with a stable structure with a constant number of side-chains free to diffuse in the effective sphere, while the increase of temperature unlocks the rotation of a higher number of methyl groups.
Finally, when cooling down the sample, although the system is now a gel, random coils undergo a partial refolding to α-helices or β-sheets. Even though refolding does not lead to the native structure, our data indicate that the dynamics of both backbone and side-chains of the non-native structure is similar to that of proteins in their native state on the nanometer length scale.
5 Conclusions
We present a comprehensive picture of hierarchical protein dynamics before and after thermal denaturation. Using high-quality data from the quasi-elastic neutron backscattering spectrometer BASIS in combination with novel and extended models for the scattering function, we provide a consistent interpretation of the data as a combination of contributions from three hierarchical dynamic levels: (i) entire-protein apparent self-diffusion, (ii) backbone fluctuations, and (iii) side-chain dynamics. As long as the protein is in its native conformation, i.e. below the transition temperature Td, the translational self-diffusion is successfully described by colloid theory for hard spheres. The dynamics of the backbone is faster at lower protein concentration, and slows down for more crowded solutions at all temperatures. Increasing temperature results in accelerated dynamics and in a larger space accessible by backbone atoms, meaning that backbone dynamics becomes less confined. The side-chain dynamics also becomes slower with increasing protein concentration, while it becomes faster with increasing T. Below Td, the geometrical confinement of the side-chains appears to be unaltered.Above Td, cross-links between the proteins and eventually a gel are formed. Connecting loops and random coils become at least as significant as intermolecular β-sheets and α-helices.32 Consequentially, the center-of-mass protein dynamics decreases dramatically. By contrast, the average backbone fluctuations become faster. While the number of side-chains that can diffuse in their local environment increases, their average motion exhibits a sharp but limited crowding-dependent slowdown during denaturation, and subsequently continues its thermally-driven acceleration. Upon re-cooling, the denatured sample remains a gel. The global apparent self-diffusion in this case is significantly lower than in the solution at the same temperature, while the internal dynamics is comparable, probably due to partial refolding of proteins, although not in their native globular state.
The association of the model parameters with the three hierarchical dynamic levels, and in particular of the two linewidths Γ1,2 with the backbone and side chain fluctuation, respectively, represents one consistent and physically justified interpretation. Although we cannot rule out other nontrivial models and interpretations of this complex system, the achieved picture of protein molecular dynamics during a complex process such as thermal denaturation is promising for future applications of quasi-elastic neutron scattering in complex systems with hierarchical dynamics down to the pico- and nanosecond time scale.
Acknowledgements
The research at Oak Ridge National Laboratory's Spallation Neutron Source was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. The spectrometer BASIS is supported by the Jülich Center for Neutron Science (JCNS), Germany, via the partner user programme and M.G. and F.Za. acknowledge travel funding by JCNS to conduct the experiments as well as a student grant by the Institut Laue–Langevin (ILL). M.H., M.G. and T.S. have obtained travel funding by ILL. We acknowledge E. Mamontov, H. Schober and S. Da Vela for fruitful discussion and R. Moody for technical assistance. We further acknowledge financial support by the DFG.References
- G. Zaccai, Science, 2000, 288, 1604–1607 CrossRef CAS PubMed.
- J. C. Whisstock and A. M. Lesk, Q. Rev. Biophys., 2003, 36, 307–340 CrossRef CAS PubMed.
- M. Karplus and J. Kuriyan, Proc. Natl. Acad. Sci., U. S. A., 2005, 102, 6679–6685 CrossRef CAS PubMed.
- B. H. Zimm and J. K. Bragg, J. Chem. Phys., 1959, 31, 526–535 CrossRef CAS.
- W. Kauzmann, Adv. Protein Chem., 1959, 14, 1–63 CrossRef CAS PubMed.
- C. Tanford, Adv. Protein Chem., 1968, 23, 121–282 CrossRef CAS PubMed.
- K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS PubMed.
- R. Lumry and H. Eyring, J. Phys. Chem., 1954, 58, 110–120 CrossRef CAS.
- C. Tanford, Adv. Protein Chem., 1970, 24, 1–95 CrossRef CAS PubMed.
- W. S. Gosal and S. B. Ross-Murphy, Curr. Opin. Colloid Interface Sci., 2000, 5, 188–194 CrossRef CAS.
- A. Clark, G. Kavanagh and S. Ross-Murphy, Food Hydrocolloids, 2001, 15, 383–400 CrossRef CAS.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed.
- J. N. Onuchic, P. G. Wolynes, Z. Luthey-Schulten and N. D. Socci, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3626–3630 CrossRef CAS.
- K. A. Dill and H. S. Chan, Nat. Struct. Biol., 1997, 4, 10–19 CrossRef CAS PubMed.
- A. V. Finkelstein and E. I. Shakhnovich, Biopolymers, 1989, 28, 1681–1694 CrossRef CAS PubMed.
- E. I. Shakhnovich and A. V. Finkelstein, Biopolymers, 1989, 28, 1667–1680 CrossRef CAS PubMed.
- P. De Gennes, J. Phys., Lett., 1977, 38, 355–358 CrossRef CAS.
- P. San Biagio, D. Bulone, A. Emanuele and M. Palma, Biophys. J., 1996, 70, 494–499 CrossRef CAS PubMed.
- M. Hennig, F. Roosen-Runge, F. Zhang, S. Zorn, M. W. Skoda, R. M. Jacobs, T. Seydel and F. Schreiber, Soft Matter, 2012, 8, 1628–1633 RSC.
- C. Le Bon, T. Nicolai and D. Durand, Macromolecules, 1999, 32, 6120–6127 CrossRef CAS.
- L. Stagg, S. Zhang, M. Cheung and W. Pernilla, Proc. Natl. Acad. Sci., U. S. A., 2007, 104, 18976–18981 CrossRef CAS PubMed.
- R. Wetzel, M. Becker, J. Behlke, H. Billwitz, S. Böhm, B. Ebert, H. Hamann, J. Krumbiegel and G. Lassmann, Eur. J. Biochem., 1980, 104, 469–478 CrossRef CAS PubMed.
- V. Lin and J. Koenig, Biopolymers, 1976, 15, 203–218 CrossRef CAS PubMed.
- J. Oakes, J. Chem. Soc., Faraday Trans. 1, 1976, 72, 228–237 RSC.
- I. Hayakawa, J. Kajihara, K. Morikawa, M. Oda and Y. Fujio, J. Food Sci., 1992, 57, 288–292 CrossRef CAS.
- A. Tobitani and S. B. Ross-Murphy, Macromolecules, 1997, 30, 4845–4854 CrossRef CAS.
- D. K. Wilkins, S. B. Grimshaw, V. Receveur, C. M. Dobson, J. A. Jones and L. J. Smith, Biochemistry, 1999, 38, 16424–16431 CrossRef CAS PubMed.
- S. Dellerue, A.-J. Petrescu, J. C. Smith and M.-C. Bellissent-Funel, Biophys. J., 2001, 81, 1666–1676 CrossRef CAS PubMed.
- A. L. Lee, K. A. Sharp, J. K. Kranz, X.-J. Song and A. J. Wand, Biochemistry, 2002, 41, 13814–13825 CrossRef CAS PubMed.
- V. Militello, V. Vetri and M. Leone, Biophys. Chem., 2003, 105, 133–141 CrossRef CAS PubMed.
- V. Militello, C. Casarino, A. Emanuele, A. Giostra, F. Pullara and M. Leone, Biophys. Chem., 2004, 107, 175–187 CrossRef CAS PubMed.
- K. Murayama and M. Tomida, Biochemistry, 2004, 43, 11526–11532 CrossRef CAS PubMed.
- F. Ding, R. K. Jha and N. V. Dokholyan, Structure, 2005, 13, 1047–1054 CrossRef CAS PubMed.
- M. Pouzot, T. Nicolai, R. Visschers and M. Weijers, Food Hydrocolloids, 2005, 19, 231–238 CrossRef CAS.
- V. Vetri, F. Librizzi, M. Leone and V. Militello, Eur. Biophys. J., 2007, 36, 717–725 CrossRef CAS PubMed.
- X.-J. Song, P. F. Flynn, K. A. Sharp and A. J. Wand, Biophys. J., 2007, 92, L43–L45 CrossRef CAS PubMed.
- A. Adel, M. Nadia, O. Mohamed and G. Abdelhafidh, Mater. Sci. Eng., C, 2008, 28, 594–600 CrossRef CAS.
- Y. Zhang, M. Lagi, D. Liu, F. Mallamace, E. Fratini, P. Baglioni, E. Mamontov, M. Hagen and S.-H. Chen, J. Chem. Phys., 2009, 130, 135101 CrossRef PubMed.
- F. Meersman, C. Atilgan, A. J. Miles, R. Bader, W. Shang, A. Matagne, B. A. Wallace and M. H. Koch, Biophys. J., 2010, 99, 2255–2263 CrossRef CAS PubMed.
- L. Fu, S. Villette, S. Petoud, F. Fernandez-Alonso and M.-L. Saboungi, J. Phys. Chem. B, 2011, 115, 1881–1888 CrossRef CAS PubMed.
- C. Corsaro and D. Mallamace, Phys. A, 2011, 390, 2904–2908 CrossRef CAS.
- N. K. Das, N. Ghosh, A. P. Kale, R. Mondal, U. Anand, S. Ghosh, V. K. Tiwari, M. Kapur and S. Mukherjee, J. Phys. Chem. B, 2014, 118, 7267–7276 CrossRef CAS PubMed.
- F. Gabel, D. Bicout, U. Lehnert, M. Tehei, M. Weik and G. Zaccai, Q. Rev. Biophys., 2003, 35, 327–367 CrossRef.
- M. Jasnin, M. Moulin, M. Haertlein, G. Zaccai and M. Tehei, Biophys. J., 2008, 95, 857–864 CrossRef CAS PubMed.
- A. Stadler, I. Digel, J. Embs, T. Unruh, M. Tehei, G. Zaccai, G. Büldt and G. Artmann, Biophys. J., 2009, 96, 5073–5081 CrossRef CAS PubMed.
- F. Roosen-Runge, M. Hennig, F. Zhang, R. M. J. Jacobs, M. Sztucki, H. Schober, T. Seydel and F. Schreiber, Proc. Natl. Acad. Sci., U. S. A., 2011, 108, 11815–11820 CrossRef CAS PubMed.
- M. Grimaldo, F. Roosen-Runge, F. Zhang, T. Seydel and F. Schreiber, J. Phys. Chem. B, 2014, 118, 7203–7209 CrossRef CAS PubMed.
- V. Receveur, P. Calmettes, J. C. Smith, M. Desmadril, G. Coddens and D. Durand, Proteins: Struct., Funct., Bioinf., 1997, 28, 380–387 CrossRef CAS.
- M. Kataoka, M. Ferrand, A. Goupil-Lamy, H. Kamikubo, J. Yunoki, T. Oka and J. Smith, Phys. B, 1999, 266, 20–26 CrossRef CAS.
- Z. Bu, D. A. Neumann, S.-H. Lee, C. M. Brown, D. M. Engelman and C. C. Han, J. Mol. Biol., 2000, 301, 525–536 CrossRef CAS PubMed.
- Z. Bu, J. Cook and D. J. Callaway, J. Mol. Biol., 2001, 312, 865–873 CrossRef CAS PubMed.
- D. Russo, J. Pérez, J.-M. Zanotti, M. Desmadril and D. Durand, Biophys. J., 2002, 83, 2792–2800 CrossRef CAS PubMed.
- M. Tarek, D. A. Neumann and D. J. Tobias, Chem. Phys., 2003, 292, 435–443 CrossRef CAS.
- J. Fitter, Biophys. J., 2003, 84, 3924–3930 CrossRef CAS PubMed.
- A. M. Gaspar, M.-S. Appavou, S. Busch, T. Unruh and W. Doster, Eur. Biophys. J., 2008, 37, 573–582 CrossRef CAS PubMed.
- H. Jansson and J. Swenson, J. Chem. Phys., 2008, 128, 245104 CrossRef CAS PubMed.
- G. Gibrat, F. Assairi, Y. Blouquit, C. Craescu and M. Bellissentfunel, Biophys. J., 2008, 95, 5247–5256 CrossRef CAS PubMed.
- M. G. Ortore, F. Spinozzi, P. Mariani, A. Paciaroni, L. R. Barbosa, H. Amenitsch, M. Steinhart, J. Ollivier and D. Russo, J. R. Soc., Interface, 2009, 6, S619 CrossRef CAS PubMed.
- E. Mamontov, H. ONeill and Q. Zhang, J. Biol. Phys., 2010, 36, 291–297 CrossRef CAS PubMed.
- S. Perticaroli, J. D. Nickels, G. Ehlers, E. Mamontov and A. P. Sokolov, J. Phys. Chem. B, 2014, 118, 7317–7326 CrossRef CAS PubMed.
- E. Mamontov and K. W. Herwig, Rev. Sci. Instrum., 2011, 82, 085109 CrossRef CAS PubMed.
- M. Grimaldo, F. Roosen-Runge, N. Jalarvo, M. Zamponi, F. Zanini, M. Hennig, F. Zhang, F. Schreiber and T. Seydel, EPJ Web conf., 2015, in press (Proceedings of QENS/WINS).
- F. Roosen-Runge, M. Hennig, T. Seydel, F. Zhang, M. Skoda, S. Zorn, R. Jacobs, M. Maccarini, P. Fouquet and F. Schreiber, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 68–75 CrossRef CAS PubMed.
- B. Jacrot, Rep. Prog. Phys., 1976, 39, 911 CrossRef CAS.
- A. Bujacz, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 1278–1289 CrossRef CAS PubMed.
- T. Seydel, L. Wiegart, F. Juranyi, B. Struth and H. Schober, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 061403 CrossRef.
- A. M. Gaspar, S. Busch, M.-S. Appavou, W. Haeussler, R. Georgii, Y. Su and W. Doster, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 76–82 CrossRef CAS PubMed.
- K. S. Singwi and A. Sjölander, Phys. Rev., 1960, 119, 863–871 CrossRef CAS.
- P. L. Hall and D. Ross, Mol. Phys., 1981, 42, 673–682 CrossRef CAS.
- F. Roosen-Runge and D. Bicout, in preparation.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- M. Tokuyama and I. Oppenheim, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1994, 50, 16–19 CrossRef.
- J. Qvist, H. Schober and B. Halle, J. Chem. Phys., 2011, 134, 144508 CrossRef PubMed.
- A. M. Stadler, C. J. Garvey, J. P. Embs, M. M. Koza, T. Unruh, G. Artmann and G. Zaccai, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 2989–2999 CrossRef CAS PubMed.
- M. Tehei, J. Perlmutter, F. Giusti, J. Sachs, G. Zaccai and J.-L. Popot, J. Membr. Biol., 2014, 247, 897–908 CrossRef CAS PubMed.
- M. Bée, Phys. B, 1992, 182, 323–336 CrossRef.
- J. Perez, J. Zanotti and D. Durand, Biophys. J., 1999, 77, 454–469 CrossRef CAS PubMed.
- F. Volino and A. Dianoux, Mol. Phys., 1980, 41, 271–279 CrossRef CAS.
- F. Volino, J.-C. Perrin and S. Lyonnard, J. Phys. Chem. B, 2006, 110, 11217–11223 CrossRef CAS PubMed.
- V. Réat, H. Patzelt, M. Ferrand, C. Pfister, D. Oesterhelt and G. Zaccai, Proc. Natl. Acad. Sci., U. S. A., 1998, 95, 4970–4975 CrossRef.
- U. Lehnert, V. Réat, M. Weik, G. Zaccai and C. Pfister, Biophys. J., 1998, 75, 1945–1952 CrossRef CAS PubMed.
- F. Mallamace, S.-H. Chen, M. Broccio, C. Corsaro, V. Crupi, D. Majolino, V. Venuti, P. Baglioni, E. Fratini, C. Vannucci and H. E. Stanley, J. Chem. Phys., 2007, 127, 045104 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp04944f |
| This journal is © the Owner Societies 2015 |