
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A functionalised nickel cyclam catalyst for CO2 reduction: electrocatalysis, semiconductor surface immobilisation and light-driven electron transfer†
Gaia
Neri‡
a,
James J.
Walsh‡
a,
Calum
Wilson
a,
Anna
Reynal
b,
Jason Y. C.
Lim
b,
Xiaoe
Li
b,
Andrew J. P.
White
b,
Nicholas J.
Long
b,
James R.
Durrant
b and
Alexander J.
Cowan
*a
aDepartment of Chemistry, Stephenson Institute for Renewable Energy, The University of Liverpool, L69 7ZF, UK. E-mail: a.j.cowan@liverpool.ac.uk
bDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK
First published on 26th November 2014
Abstract
The immobilisation of electrocatalysts for CO2 reduction onto light harvesting semiconductors is proposed to be an important step towards developing more efficient CO2 reduction photoelectrodes. Here, we report a low cost nickel cyclam complex covalently anchored to a metal oxide surface. Using transient spectroscopy we validate the role of surface immobilisation on enhancing the rate of photoelectron transfer. Furthermore [Ni(1,4,8,11-tetraazacyclotetradecane-6-carboxylic acid)]2+ (2) is shown to be a very active electrocatalyst in solution.
The photoelectrochemical reduction of CO2 to products such as carbon monoxide, formic acid and methanol is receiving intense interest.1,2 When coupled to a water oxidation photocatalyst, light driven CO2 reduction offers a sustainable route to carbon-based fuels from renewable feedstock. However, to date, the efficiency of such approaches has remained unfeasibly low and significant challenges remain including high electron–hole recombination yields, low selectivities towards CO2 and the use of high cost materials and solvents.
Photoelectrochemical reduction of CO2 in water is particularly challenging, as competitive proton reduction occurs at similar potentials.3 A promising approach to obtaining higher selectivity towards CO2 reduction over H2 production and increased solar to fuel efficiencies is to introduce a molecular electrocatalyst with a high selectivity towards CO2.4 Numerous studies have explored the use of photocathodes to drive electrocatalysis in solution.5 Immobilisation of the electrocatalyst directly onto the photocathode surface to form a hybrid photoelectrode is also receiving increasing interest due to anticipated improvements in the rate of charge transfer to the molecular catalyst and in the stability and recyclability of the system.6 Immobilisation of a molecular catalyst either within a polymer or by electropolymerisation has been explored in several studies,7,8 including using [Re(bpy)(CO)3Br] and [Co(bpy)3]2+, with marked improvements in photoelectrochemical activity towards CO2 being achieved. A series of studies9,10 explored the electropolymerisation of ruthenium electrocatalysts onto InP and GaP photoelectrodes with an overall solar to fuel efficiency of 0.14% being reported for a SrTiO3/InP/Ru device for the production of formate from water and CO2. In contrast to the number of polymer-immobilised systems, few examples of directly anchored molecular catalysts are known, despite offering potential advantages over the control of the catalyst binding modes, which is highly desirable for optimising charge transfer at the semiconductor/catalyst interface. In 2010, Sato et al.6,11 reported the immobilisation of a ruthenium-based molecular electrocatalyst modified with carboxylic acid or phosphonic acid binding groups onto p-type N–Ta2O5 for use as a photocatalyst in CH3CN. Significantly this study showed that the photocatalytic activity of the hybrid material far exceeded that of the two-components simply mixed in solution, with related transient absorption spectroscopy (TAS) measurements identifying charge transfer from the semiconductor to the immobilised electrocatalyst as a potentially significant factor.12 Recently Ishitani et al. reported that [Ru{4,4′-(CH2PO3H2)2-2,2′-bipyridine}(CO)2Cl2] immobilised on g-C3N4 is an active photocatalyst for reducing CO2 to formic acid with a selectivity of >80% and a turnover number (TON) of >200.13 The same group has also explored the binding of ruthenium catalysts to TaON for CO2 reduction.14
To the best of our knowledge, all of the examples of directly immobilised CO2 molecular electrocatalysts on light absorbing semiconductors have either employed high cost metal centres (e.g. Ru)6 or complete enzymes.15 Here we explore the covalent immobilisation of a low cost [Ni(cyclam)]2+ (cyclam = 1,4,8,11-tetraazacyclotetradecane, (1)) derivative to metal oxide photoelectrodes. [Ni(cyclam)]2+ and its derivatives are widely studied electrocatalysts due to their high stability and selectivity towards the reduction of CO2 to CO in water on mercury electrodes.16–19 Furthermore this class of catalyst is weakly coloured (εd–d ∼ 10–50), thereby avoiding detrimental inner-filter effects when used as the catalyst in a sensitised photocatalytic system. Photocatalytic CO2 reduction has been reported for solutions containing 1 and a molecular photosensitiser such as [Ru(bpy)3]2+ (where bpy = 2,2′-bipyridyl),20,21 and for supramolecular systems consisting of ruthenium polypyridyl complexes covalently linked to 1.22,23 [Ni(cyclam)]2+ has also been used in solution with a range of p-type photoelectrodes such as p-Si, p-GaP or p-GaAs,24–26 which demonstrates the viability of using derivatives of 1 as catalysts in a light-driven system.
The synthetic procedure for the dichloride salt of 2, a [Ni(cyclam)]2+ complex modified with a carboxylic acid group for binding to metal-oxide surfaces, is described in the ESI.† We identified functionalisation of the carbon backbone as an appropriate route as it is known that the presence of the quaternary N–H protons within 1 are critical in aiding CO2 binding and catalysis, with functionalisation of the amine groups leading to decreased selectivity.27,28 The single crystal X-ray structure of the complex (Fig. 1) shows the nickel centre to have a slightly distorted octahedral coordination geometry (cis angles in the range 85.51(8)° to 95.01(8)°), with the two chlorine ligands occupying the axial sites. In the crystal, the complex assumes a R,R,S,S (trans-III) conformation.29 In aqueous solution, 1 primarily adopts a square planar configuration; however, in contrast, UV/Vis studies of 2 (Fig. S8, ESI†) indicate the presence of a mix of octahedral and square planar geometries in aqueous solutions.30 In line with these observations, the 1H-NMR for complex 2 in D2O presents a mix of very broad and resolved peaks, characteristic of species exhibiting a degree of paramagnetism.
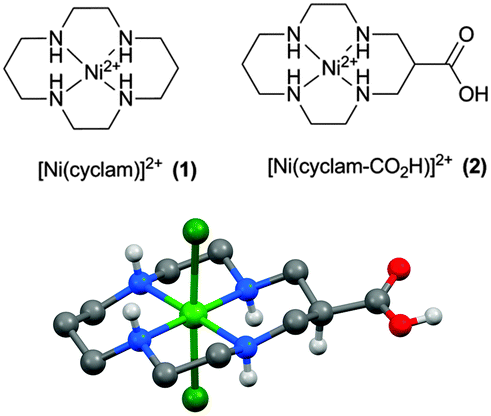 | ||
| Fig. 1 (a) [Ni(cyclam)]2+ (1) was functionalised on the carbon backbone with a carboxylic acid group to yield (2) for anchoring to metal oxide electrodes. (b) X-ray structure of 2. H atoms, except for those bound to N or O, are emitted for clarity (details in ESI†). | ||
A very small number of studies have demonstrated that by careful control of the complex geometry it is possible to develop nickel cyclam derivative CO2 reduction electrocatalysts with higher turnover frequencies and lower onset potentials;17,18,31 however more typically cyclam modification is found to have significant detrimental effects on the electrocatalytic activity towards CO2. Therefore it is interesting to also assess the electrocatalytic activity of 2 in solution. Cyclic voltammograms measured in 0.1 M aqueous NaClO4 at pH 5 on a hanging mercury drop electrode (HMDE) are shown in Fig. 2 and Fig. S4–S6 (ESI†). On the HMDE the NiII/I couple of 2 under argon appears at −1.55 V vs. Ag/AgCl, close to that of the unmodified complex 1 (−1.50 V vs. Ag/AgCl, inset in Fig. 2a),16 and a reversible NiII/III couple for 2, studied under argon on a glassy carbon working electrode at 0.43 V (vs. Fc/Fc+, ca. 0.8 V vs. Ag/AgCl) is also noted (Fig. S1–S3, ESI†). Under CO2 a large increase in current is observed for both 1 and 2 at potentials close to that of NiII/I under argon. In-line with the known activity of 1, this is assigned to the electrocatalytic reduction of CO2 to CO.16 Bulk electrolysis measurements carried out at −1.4 V (vs. Ag/AgCl) using an Hg–Au amalgam working electrode and 10−4 M solutions of 2 in 0.1 M NaClO4 confirm that the large current enhancement under CO2 is due to electrocatalytic CO2 reduction to CO. An excellent selectivity towards CO2 reduction is achieved with a CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 product ratio of ∼100:1 as measured by headspace gas chromatography, with a combined Faradaic efficiency of 88% and a turnover number for CO2 reduction of 5.3 within 1 hour, indicating that modification of the carbon backbone of the cyclam ligand has not notably decreased the catalytic activity of 2 (Fig. S7, ESI†). To further assess the electrocatalytic activity of 2 we have measured the ratio of the peak currents in the absence (ip) and presence (ipc) of the CO2 substrate, which is a commonly used method to estimate electrocatalytic activity. Here we estimate ipc/ip ∼ 48 for complex 2 which is slightly greater than the value for 1 (ipc/ip ∼ 31). This could be taken to indicate enhanced catalytic activity, however it is important to note that the electrocatalysis onset is ∼50 mV more cathodic for 2 (−1.27 V) than 1 (−1.22 V) and that accurate determination of ip for 2 is complicated by the onset of hydrogen reduction at these negative potentials. Instead, in-line with a previous study on related complexes,18 a Tafel analysis was employed to provide a more detailed analysis of the electrocatalytic activity of 2 (Fig. 3). Analysis of the slow scan rate (2 mV s−1) linear sweep voltammograms (LSVs) for 2 reveals that a significant pre-wave exists at overpotentials less than −0.6 V. Similar pre-waves have been observed for other modified nickel cyclams and they correlate to the adsorption and geometric reorganisation of the catalyst.18 The slopes of the Tafel plots for 2 and 1 are found to be indistinguishable (56 mV per decade) for overpotentials between −0.61 and −0.99 V, the region confirmed where CO2 reduction occurs (Fig. S7, ESI†), further reinforcing that functionalisation of the cyclam structure has not detrimentally altered the electrocatalytic activity on mercury at pH 5.
:H2 product ratio of ∼100:1 as measured by headspace gas chromatography, with a combined Faradaic efficiency of 88% and a turnover number for CO2 reduction of 5.3 within 1 hour, indicating that modification of the carbon backbone of the cyclam ligand has not notably decreased the catalytic activity of 2 (Fig. S7, ESI†). To further assess the electrocatalytic activity of 2 we have measured the ratio of the peak currents in the absence (ip) and presence (ipc) of the CO2 substrate, which is a commonly used method to estimate electrocatalytic activity. Here we estimate ipc/ip ∼ 48 for complex 2 which is slightly greater than the value for 1 (ipc/ip ∼ 31). This could be taken to indicate enhanced catalytic activity, however it is important to note that the electrocatalysis onset is ∼50 mV more cathodic for 2 (−1.27 V) than 1 (−1.22 V) and that accurate determination of ip for 2 is complicated by the onset of hydrogen reduction at these negative potentials. Instead, in-line with a previous study on related complexes,18 a Tafel analysis was employed to provide a more detailed analysis of the electrocatalytic activity of 2 (Fig. 3). Analysis of the slow scan rate (2 mV s−1) linear sweep voltammograms (LSVs) for 2 reveals that a significant pre-wave exists at overpotentials less than −0.6 V. Similar pre-waves have been observed for other modified nickel cyclams and they correlate to the adsorption and geometric reorganisation of the catalyst.18 The slopes of the Tafel plots for 2 and 1 are found to be indistinguishable (56 mV per decade) for overpotentials between −0.61 and −0.99 V, the region confirmed where CO2 reduction occurs (Fig. S7, ESI†), further reinforcing that functionalisation of the cyclam structure has not detrimentally altered the electrocatalytic activity on mercury at pH 5.
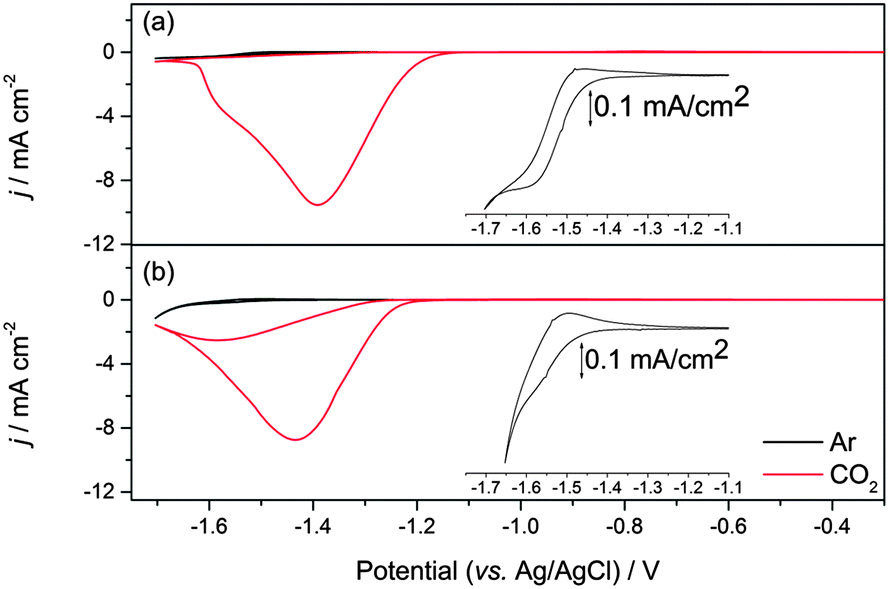 | ||
| Fig. 2 CV of ca. 1 mM solutions of 1 (a) and 2 (b) recorded in 0.1 M NaClO4 at pH 5 purged with either argon (black line) or CO2 (red line) recorded at 100 mV s−1 using a HMDE electrode (0.023 cm−2). The inset shows an expansion of the NiII/I couple under argon, see also Fig. S4 (ESI†). | ||
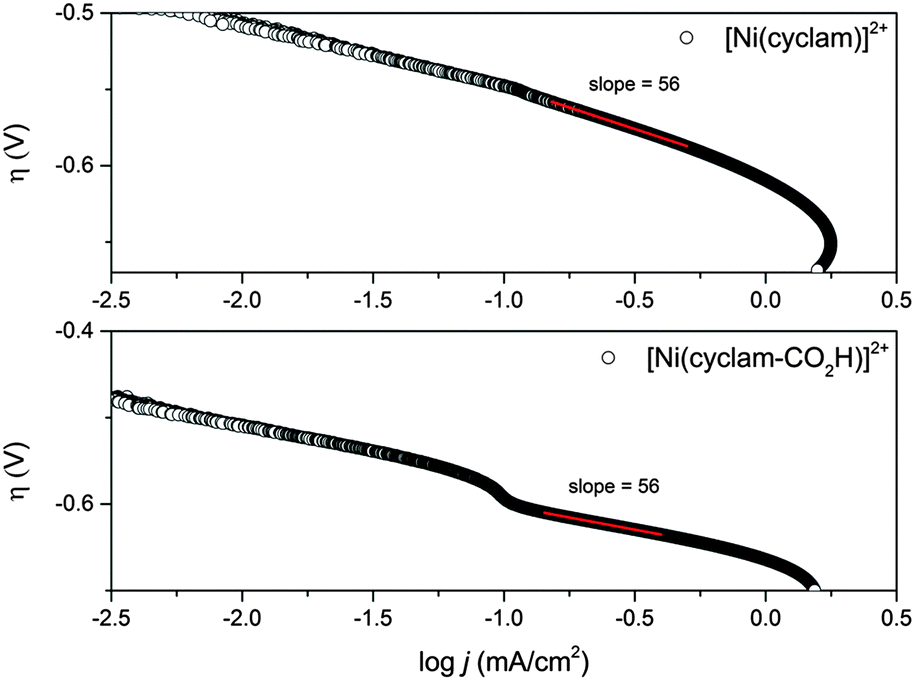 | ||
| Fig. 3 Plots of CO2 reduction overpotential (pH = 5) vs. log of current density for catalysts 1 and 2. Calculated from LSVs at 2 mV s−1 in 0.1 M NaClO4 electrolyte containing 1 × 10−4 M catalyst. Overpotentials were calculated from the thermodynamic potential for CO2 reduction at pH = 5 (−0.41 V vs. NHE).18 | ||
The presence of a suitable binding group on 2 permits the immobilisation of this catalyst onto metal oxide electrodes. Binding of 2 to semiconductor surfaces was performed by dip-coating nanocrystalline (nc) TiO2 films (particle ϕ ∼ 20 nm, film 3 μm thick) on FTO glass in a 2 mM ethanolic solution of the catalyst for 48 hours. Catalyst uptake was evaluated and quantified by both measuring the decrease in absorbance of 2 in the soaking solution using UV-vis spectroscopy and through desorption of 2 from the surface using 1 M NaOH; typical experiments resulted in approximately 1000 molecules of 2 bound per TiO2 nanoparticle (see ESI† for calculations). The binding mode of 2 to nc-TiO2 films was examined using FTIR spectroscopy (Fig. S9, ESI†), which showed a clear shift in the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) frequency of the carboxylic acid group of 2 when compared to that of the unbound solid sample. Careful analysis of the splitting of the asymmetric and symmetric stretching modes of the carboxylate group indicates that 2 is likely to be bound in a monodentate manner through this group to the TiO2 surface.
O) frequency of the carboxylic acid group of 2 when compared to that of the unbound solid sample. Careful analysis of the splitting of the asymmetric and symmetric stretching modes of the carboxylate group indicates that 2 is likely to be bound in a monodentate manner through this group to the TiO2 surface.
High surface area n-type semiconductors such as nc-TiO2 have been explored as supports for reductive electrochemistry in the dark.32 In contrast to traditional conductive electrodes the redox species of interest is only observed at potentials close to, or negative of, the conduction band edge, i.e. when the semiconductor is no longer acting as an insulator.32 The energy of the TiO2 conduction band (CB) edge is dependent upon both the solvent and the nature of the electrolyte ions studied.33 In very dry aprotic solvents such as CH3CN with (But)4NPF6 the CB potential is ca. −2.1 V (vs. Ag/AgCl),33 sufficiently negative of the solution NiII/I potential of complex 2. Here we have examined the electrochemistry of both 1 in solution with a TiO2 electrode and of 2 immobilised on a TiO2 electrode in CH3CN. In the absence of a catalyst the CV of nc-TiO2 in CH3CN shows behaviour associated with the charging and discharging of trap states close to the conduction band edge, in line with previous reports, Fig. 4.34,35 In contrast the electrochemical response of nc-TiO2–2 is markedly different with the presence of a new reductive feature at −1.5 V (vs. Fc/Fc+), assigned to the reduction of the NiII of complex 2, indicating that the catalyst remains electrochemically active on the semiconductor surface and that electron transfer from the semiconductor to the catalyst is achievable. The dependence of the current response on increasing scan rate (Fig. S12, ESI†) indicates the absence of diffusion contributions, in line with a surface bound species. Control experiments using a blank nc-TiO2 electrode with either 1 or 2 in solution showed no features assignable to the NiII/I redox couple (Fig. S10 and S11, ESI†). Addition of CO2 to the nc-TiO2–2 system does lead to an initial increase in cathodic current, which may indicate electrocatalytic activity (Fig. S13, ESI†), however the system is found to be insufficiently stable under CO2 for bulk electrolysis experiments. Interestingly we also find that the potential of the NiII/I reduction of 2 on TiO2 in CH3CN (−1.4 V vs. Fc/Fc+, ca. −1.1 vs. Ag/AgCl) is shifted anodically compared with on a mercury electrode at pH 5 under argon, demonstrating a potential advantage of the immobilised system.
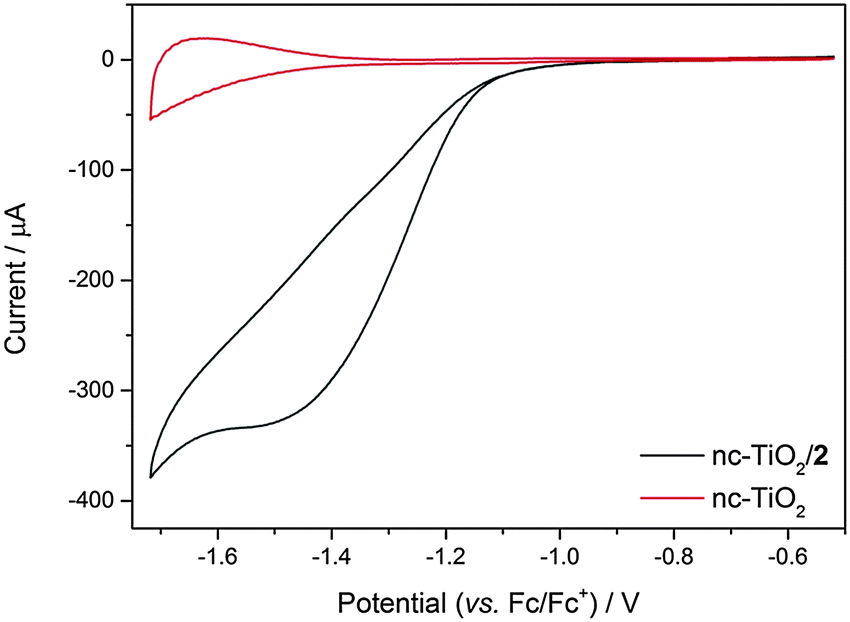 | ||
| Fig. 4 Cyclic voltammograms of nc-TiO2 (red) and nc-TiO2–2 (black) under argon in 0.1 M (But)4NPF6/MeCN. v = 100 mV s−1. | ||
As the yield of photoelectron transfer across the catalyst/semiconductor interface is likely to be a critical factor in determining overall photocatalytic efficiency, it is important that the fundamental design rules controlling electron transfer to immobilised catalysts are explored. In order to identify if photoinduced electron transfer can occur from the semiconductor electrode to the immobilised cyclam complex 2, we have studied TiO2–2 in deaerated CH3CN in the presence of triethanolamine (TEOA, 0.1 M) using TAS. TAS has been widely used to study the dynamics of photoelectrons and holes in TiO2 and it is known that conduction band photoelectrons have an absorption feature at wavelengths greater than 800 nm.36 Following direct band gap excitation (355 nm, 6 ns) of nc-TiO2 in the absence of either 1 or 2 with TEOA as the hole scavenger we observe a very long-lived transient absorption signal at 900 nm, that decays with t50% = 0.8 s, Fig. 5. This TAS signal is assigned to long-lived TiO2 photoelectrons, as electron–hole recombination processes are suppressed due to the rapid scavenging of holes by TEOA.37 In contrast, TAS experiments on TiO2–2 recorded under identical conditions show a rate of photoelectron decay (t50% = 1.2 ms) that is over two orders of magnitude faster than the control experiment of bare TiO2. The rapid decay of the TiO2 photoelectron signal indicates that efficient electron transfer from the TiO2 to the immobilised catalyst occurs, in good agreement with our earlier electrochemical studies that demonstrated electron transfer from the conduction band of TiO2 to the NiII/I couple is thermodynamically viable (Fig. 4). We have also carried out experiments of unmodified TiO2 electrodes with complex 1 (ca. 1 × 10−4 M) in solution to explore the role of surface immobilisation on electron transfer kinetics and these data reveal that electron transfer from the metal oxide to [Ni(cyclam)]2+ in solution is an order of magnitude slower (t50% = 20 ms) than that observed for the hybrid TiO2–2 system, confirming that direct covalent immobilisation markedly improves the rate of photoelectron transfer to the electrocatalyst.
 | ||
| Fig. 5 Transient absorption decays of (a) unmodified TiO2 (black), TiO2–2 (blue) and TiO2 with 1 10−4 M in solution (green) and (b) Ti1−xZrO2 (black) and Ti1−xZrO2–2 (blue) measured in CH3CN with 0.1 M TEOA. The samples were excited at 355 nm (350 μJ cm−2) and probed at 900 nm. The samples were purged under N2 for 15 min prior to measurements. A useful measure for the kinetics of the process is t50% (dotted lines), which represents the time delay by which the yield of photoelectrons has dropped by 50% when compared to an initial concentration, here defined to be the photoelectron yield at 4 microseconds. | ||
A very recent study on water splitting systems has shown that a key parameter controlling the rate of photoelectron transfer is the distance between the semiconductor and the catalytic core.37 It is however also important to understand the role of the thermodynamic driving force on the rate of photoelectron transfer. Here we have also immobilised complex 2 on a mixed Ti1−xZrxO2 film (x = 0.2, see Fig. S14 and S15, ESI†). It has been previously shown that Ti1−xZrxO2 has a conduction band edge that is shifted by ca. 150 mV vs. nc-TiO2 and this is confirmed through spectroelectrochemical measurements, Fig. S16 (ESI†). TAS measurements on Ti1−xZrxO2–2 show a marked increase in the rate of photoelectron decay (t50% = 800 μs) when compared to a Ti1−xZrxO2 film in the absence of the catalyst (t50% = 500 ms), indicating that photoelectron transfer to 2 from Ti1−xZrxO2 is occurring (Fig. S17, ESI†). The faster rate of electron transfer in Ti1−xZrxO2–2 is in-line with the increased thermodynamic driving force for electron transfer (∼0.65 eV, Table S1, ESI†) when compared to TiO2–2 (∼0.5 eV, t50% = 1.2 ms), indicating that optimisation of the driving force for electron transfer to the catalyst is also an important parameter for achieving efficient charge transfer.
Conclusions
Here we report on the immobilisation of a low-cost nickel cyclam derivative onto semiconductor materials with potential applications for high surface area electrochemistry and photoelectrochemical CO2 reduction. To the best of our knowledge this represents the first example of photo-driven electron transfer to nickel cyclam covalently anchored to a semiconductor surface. Initial studies have indicated that 2 is a promising electrocatalyst for immobilisation as it is able to accept photoelectrons from semiconductor materials including TiO2 and Ti1−xZrO2 in aprotic solvents. However issues regarding stability need to be addressed for stable photoelectrochemical CO2 reduction to occur in protic solvents; a promising approach may be to explore secondary polymer encapsulation as this is known to enhance the stability of immobilised coordination compounds on TiO2 without a detrimental effect on the electrical properties.38 A key component of this study is a fundamental assessment of the factors controlling charge transfer to molecular electrocatalysts which confirms the effectiveness of the approach of hybrid covalently immobilised molecular–semiconductor system for enabling efficient photoelectron transfer.Acknowledgements
AJC and JJW acknowledge the EPSRC for a fellowship (EP/K006851/1) and financial support respectively, Prof. K. Durose and Dr R. Treharne for access to the profilometer, Dr T. D. Veal and Mr M. Birkett for use of the FTIR. Prof. M. Brust is thanked for the loan of the HMDE. AR thanks the European Commission Marie Curie CIG (PhotoCO2). JRD, AR and LX thank the ERC project Intersolar for funding.Notes and references
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- W. Tu, Y. Zhou and Z. Zou, Adv. Biomater., 2014, 26, 4607–4626 CAS.
- T. Yui, Y. Tamaki, K. Sekizawa and O. Ishitani, in Photocatalysis, ed. C. A. Bignozzi, Springer, Berlin, Heidelberg, 2011, vol. 303, pp. 151–184 Search PubMed.
- B. Kumar, J. M. Smieja and C. P. Kubiak, J. Phys. Chem. C, 2010, 114, 14220–14223 CAS.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 CrossRef CAS PubMed.
- S. Chardon-Noblat, M. N. Collomb-Dunand-Sauthier, A. Deronzier, R. Ziessel and D. Zsoldos, Inorg. Chem., 1994, 33, 4410–4412 CrossRef CAS.
- T. Hirose, Y. Maeno and Y. Himeda, J. Mol. Catal. A: Chem., 2003, 193, 27–32 CrossRef CAS.
- T. Arai, S. Sato, K. Uemura, T. Morikawa, T. Kajino and T. Motohiro, Chem. Commun., 2010, 46, 6944–6946 RSC.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274–1282 CAS.
- T. M. Suzuki, H. Tanaka, T. Morikawa, M. Iwaki, S. Sato, S. Saeki, M. Inoue, T. Kajino and T. Motohiro, Chem. Commun., 2011, 47, 8673–8675 RSC.
- K.-i. Yamanaka, S. Sato, M. Iwaki, T. Kajino and T. Morikawa, J. Phys. Chem. C, 2011, 115, 18348–18353 CAS.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127–10129 RSC.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef CAS PubMed.
- A. Bachmeier, S. Hall, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2014, 136, 13518–13521 CrossRef CAS PubMed.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316 RSC.
- E. Fujita, J. Haff, R. Sanzenbacher and H. Elias, Inorg. Chem., 1994, 33, 4627–4628 CrossRef CAS.
- J. Schneider, H. Jia, K. Kobiro, D. E. Cabelli, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2012, 5, 9502–9510 CAS.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef CAS PubMed.
- J. L. Grant, K. Goswami, L. O. Spreer, J. W. Otvos and M. Calvin, J. Chem. Soc., Dalton Trans., 1987, 2105–2109 RSC.
- C. A. Craig, L. O. Spreer, J. W. Otvos and M. Calvin, J. Phys. Chem., 1990, 94, 7957–7960 CrossRef CAS.
- E. Kimura, X. H. Bu, M. Shionoya, S. J. Wada and S. Maruyama, Inorg. Chem., 1992, 31, 4542–4546 CrossRef CAS.
- C. Herrero, A. Quaranta, S. El Ghachtouli, B. Vauzeilles, W. Leibl and A. Aukauloo, Phys. Chem. Chem. Phys., 2014, 16, 12067–12072 RSC.
- J.-P. Petit, P. Chartier, M. Beley and J.-P. Deville, J. Electroanal. Chem. Interfacial Electrochem., 1989, 269, 267–281 CrossRef CAS.
- M. G. Bradley and T. Tysak, J. Electroanal. Chem., 1982, 135, 153–157 CrossRef CAS.
- J. P. Petit, P. Chartier, M. Beley and J. P. Deville, J. Electroanal. Chem., 1989, 269, 267–281 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932–3934 CrossRef CAS PubMed.
- K. Bujno, R. Bilewicz, L. Siegfried and T. A. Kaden, J. Electroanal. Chem., 1998, 445, 47–53 CrossRef CAS.
- B. Bosnich, M. L. Tobe and G. A. Webb, Inorg. Chem., 1965, 4, 1109–1112 CrossRef CAS.
- A. Anichini, L. Fabbrizzi, P. Paoletti and R. M. Clay, Inorg. Chim. Acta, 1977, 24, L21–L23 CrossRef CAS.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- S. N. Frank and A. J. Bard, J. Am. Chem. Soc., 1975, 97, 7427–7433 CrossRef CAS.
- G. Redmond and D. Fitzmaurice, J. Phys. Chem., 1993, 97, 1426–1430 CrossRef CAS.
- E. Topoglidis, T. Lutz, J. R. Durrant and E. Palomares, Bioelectrochemistry, 2008, 74, 142–148 CrossRef CAS PubMed.
- E. Topoglidis, A. E. G. Cass, B. O'Regan and J. R. Durrant, J. Electroanal. Chem., 2001, 517, 20–27 CrossRef CAS.
- D. Bahnemann, A. Henglein, J. Lilie and L. Spanhel, J. Phys. Chem., 1984, 88, 709–711 CrossRef CAS.
- A. Reynal, J. Willkomm, N. M. Muresan, F. Lakadamyali, M. Planells, E. Reisner and J. R. Durrant, Chem. Commun., 2014, 50, 12768–12771 RSC.
- K.-R. Wee, M. K. Brennaman, L. Alibabaei, B. H. Farnum, B. Sherman, A. M. Lapides and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 13514–13517 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental protocols, prolonged CPE, FTIR and UV/vis of films. CCDC 1028579. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cp04871g |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2015 |