
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon nanotubes oxidized by a green method as efficient metal-free catalysts for nitroarene reduction†
Shuchang
Wu
a,
Guodong
Wen
a,
Robert
Schlögl
b and
Dang Sheng
Su
*a
aShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, 72 Wenhua Road, Shenyang 110016, P. R. China. E-mail: dssu@imr.ac.cn; Fax: +86-24-8397-0019; Tel: +86-024-23971577
bDepartment of Inorganic Chemistry, Fritz Harber Institute of the Max Planck Society, Faradayweg 4-6, Berlin, 14195, Germany
First published on 26th November 2014
Abstract
Hydrogen peroxide (H2O2) functionalized carbon nanotubes exhibited better catalytic performance than their nitric acid oxidized counterparts in the reduction of nitrobenzene. One important reason may be attributed to the notably less negative oxygenated groups on the surface of the former one.
Carbon is one of the most abundant materials on the earth, and has found applications in various fields, including catalysis.1 The direct use of carbon as an efficient and low cost catalyst is an attractive topic and meets the requirements of green and sustainable chemistry. Carbon, especially nanocarbon, was proved to be an excellent catalyst for the oxidative dehydrogenation (ODH) of both ethylbenzene and other light alkanes.2–4 Besides, carbon materials such as graphite oxide, graphene and carbon nanotubes (CNTs) are suitable candidates for many liquid phase reactions. For example, benzyl alcohol could be selectively converted to its corresponding aldehyde in the presence of graphite oxide or nanoshell carbon;5,6 the oxidative coupling of benzylamine was promising in the presence of graphite oxide or doped graphene,7–9 the nitrogen doped CNTs performed even better than metal or metal oxide for cyclohexane oxidation.10
Selective reduction of nitroarenes is an important organic reaction, because the products are important intermediates for industrial applications.11 The commonly used catalysts were based on different metals or metal oxides.12–15 However, carbon could also serve as a satisfactory metal-free catalyst for this reaction.16–18 We found that the carbon nanotube oxidized by nitric acid (oCNT) performed better than the non-oxidized sample, and the performance could be further notably improved if the oCNT was annealed under noble gas flow at a certain temperature. Further studies suggested that the surface oxygenated groups were critical to activity. Among various functionalities, the carbonyl group seemed to be the most important, while the carboxylic group and anhydride played negative roles.19,20 In addition, the roles of different oxygenated functionalities were also studied by using various model catalysts. The results confirmed that the nitrobenzene reduction could occur under metal free conditions, and the oxygen functional groups played different roles.21 These findings shed light on designing more effective catalysts for selective reduction of nitroarenes.
Oxidation is a powerful method for functionalizing CNTs with oxygen-containing groups. Although the CNT could be effectively functionalized with flourishing oxygenated groups by concentrated nitric acid and/or its mixture with sulfuric acid under harsh conditions,22–24 the corrosive acids as well as the nitrogen oxides generated during oxidation made this method environmentally unfriendly. In addition, thermal treatment of the oCNT was necessary to improve the activity as far as nitrobenzene reduction was concerned,19,20 leading to the consumption of extra energy. Hydrogen peroxide (H2O2) is a green oxidant, and is used in many organic reactions. In fact, it could also be used to functionalize CNTs under mild conditions.25,26 Herein, the CNT was oxidized by H2O2 under different conditions and the obtained materials were employed as metal-free catalysts in the reduction of nitroarenes. It was shown that the H2O2 functionalized samples exhibited better catalytic performance than that modified by nitric acid. These findings may provide a facile procedure to fabricate efficient metal-free catalysts for sustainable chemistry.
In order to prepare different carbon catalysts, the commercially available CNT was first treated with concentrated HCl at room temperature and was denoted as rCNT. The oCNT was obtained by oxidizing the rCNT with concentrated HNO3 in a 100 °C oil bath for 4 h. To fabricate the diluted H2O2 functionalized catalyst, 1 g of the rCNT was dispersed in 20 mL of distilled water in an autoclave, followed by the addition of 4 mL of 30% H2O2 into the mixture. The autoclave was then sealed and kept in a 100 °C oven for 4 h, and the resulting sample was designated as CNT-HP.
The catalytic performances of different samples were tested for the reduction of nitrobenzene using hydrazine monohydrate as the reducing agent. When the reaction was carried out in the presence of CNT-HP, the reaction proceeded quite fast at the initial stage: nearly 50% of nitrobenzene was converted within only 1 h. Then the reaction slowed down, but after 4 h only a small amount of nitrobenzene remained, and the substrate could not be detected after 5 h (Fig. 1). In addition, an excellent linear relationship between the initial conversion and the CNT-HP loading was observed (Fig. S1, ESI†). Aniline was the dominant product during the whole process, and its selectivity was maintained at about 95% (Fig. S2a, ESI†). In comparison, if the oCNT was employed as the catalyst, only 74.2% of nitrobenzene conversion was observed within 4 h, and there was still about 10% of nitrobenzene left after 5 h, suggesting that the oCNT was not as active as CNT-HP. But the main product was still aniline, and its selectivity was also around 95% (Fig. S2b, ESI†). The experiments were repeated from two different aspects. On the one hand, we tested the efficiency of CNT-HP and oCNTs for nitrobenzene reduction in six parallel reactions, and each time fresh material derived from the same source was used (Fig. S3, ESI†). On the other hand, we studied the catalytic performance of CNT-HP and oCNTs in four different sets for the nitrobenzene reduction, in which fresh materials from different sources were used (Fig. S4, ESI†). All of the results indicated that H2O2 oxidation may be an efficient method to prepare highly active carbon catalysts for nitrobenzene reduction.
 | ||
| Fig. 1 Reduction of nitrobenzene catalyzed by different CNT based catalysts. Reaction conditions: 20 mg of catalyst, 1.2 g of nitrobenzene, 5.0 equivalent of hydrazine monohydrate, 95 °C. | ||
Since H2O2 is a green reagent, the oxidation of CNTs by H2O2 may be promising for certain utilization. To prepare more active carbon catalysts, we oxidized rCNTs with concentrated H2O2 under ambient atmosphere at different temperatures. Specifically, 1 g of rCNT was dispersed in 25 mL of 30% H2O2, and treated at different temperatures. The obtained material was denoted as CNT-HPX (X was the oxidation temperature) and employed in the reduction of nitrobenzene. Fig. 2 showed that the catalytic performance improved remarkably compared to CNT-HP when the rCNT was treated with concentrated H2O2. Each of the modified catalyst would provide a good catalytic result even at a relatively low reaction temperature (70 °C). In comparison, only 46.6% of nitrobenzene conversion was achieved for CNT-HP under otherwise the same reaction conditions.
 | ||
| Fig. 2 Reduction of nitrobenzene catalyzed by different H2O2 oxidized CNT samples. Reaction conditions: 20 mg of catalyst, 1.2 g of nitrobenzene, 5.0 equivalent of hydrazine monohydrate, 70 °C, 4 h. | ||
We then examined the scope of substrates for the carbon-catalyzed reduction. The conversion of different nitroarenes, such as 1,3-dinitrobenzene, 4-nitroanisole and 2-nitrofluorene were tested under the optimized reaction conditions in the presence of CNT-HP60. The results illustrated that full conversion was observed for each of the tested substrate, with good to excellent selectivity for the corresponding aniline (Table 1). These findings suggested that the functionalized CNTs could not only effectively catalyze the reduction of nitrobenzene, but also serve as good catalysts for the reduction of some substituted nitroarenes.
| Entry | Substrate | Product | Conv. (%) | Sel. (%) |
|---|---|---|---|---|
| a 20 mg of the catalyst, 5 mmol of the substrate, 5 equivalent of hydrazine monohydrate, 100 °C, 5 h. b 20 mg of the catalyst, 5 mmol of the substrate, n(N2H4)/n(–NO2) = 5, 2 mL of ethanol, 100 °C, 5 h. c 50 mg of the catalyst, 5 mmol of the substrate, 5 equivalent of hydrazine monohydrate, 2 mL of ethanol, 70 °C, 4 h. d 200 mg of the catalyst, 5 mmol of the substrate, 5 equivalent of hydrazine monohydrate, 2 mL of ethanol, 70 °C, 4 h. e 100 mg of the catalyst, 5 mmol of the substrate, 5 equivalent of hydrazine monohydrate, 5 mL of ethanol, 70 °C, 10 h. | ||||
| 1a |
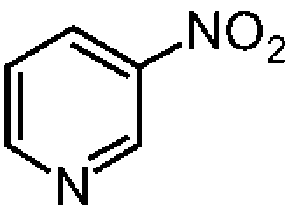
|

|
99.5 | 86.7 |
| 2b |
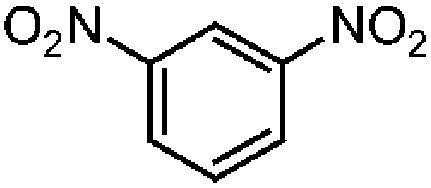
|

|
100 | 92.3 |
| 3c |

|

|
100 | 99.6 |
| 4c |

|

|
100 | 93.9 |
| 5d |

|

|
100 | 98.7 |
| 6e |

|

|
100 | 100 |
To elucidate the reason for the improved performance of H2O2 oxidized CNTs compared to oCNTs, the catalysts were studied by transmission electron microscopy (TEM), N2 adsorption, thermogravimetric analysis (TGA), X-ray photoelectron spectroscopy (XPS) and temperature programmed desorption (TPD). The TEM characterization was used to find if the different oxidation methods had a notable effect on the morphology of the carbon material. The TEM images (Fig. 3) revealed that the CNT samples existed in tangled form, while high resolution TEM showed that there was a small amount of amorphous carbon on the surface of each catalyst. So the morphologies of the catalysts were not changed no matter which oxidation conditions were selected in this study, and the improved activity of CNT-HP60 compared to other catalysts was not likely to have originated from the morphology aspect.
 | ||
| Fig. 3 TEM images of different CNT catalysts. | ||
The specific surface area was measured by the Brunauer–Emmett–Teller (BET) method using nitrogen adsorption–desorption isotherms. The BET specific surface area was 195.7 m2 g−1 for rCNT, while the values were 199.6, 205.4 and 211.3 m2 g−1 for CNT-HP, CNT-HP60 and oCNT, respectively, suggesting that different oxidation methods has no obvious effect on the surface area. In addition, these catalysts exhibited nearly the same hysteresis loops (Fig. S5, ESI†). The TGA was performed under a flow of argon (50 mL min−1) with a heating rate of 10 °C min−1 from 35 to 950 °C. The results revealed that oCNTs may contain the highest amount of oxygen functional groups, followed by CNT-HP60 and CNT-HP (Fig. 4).
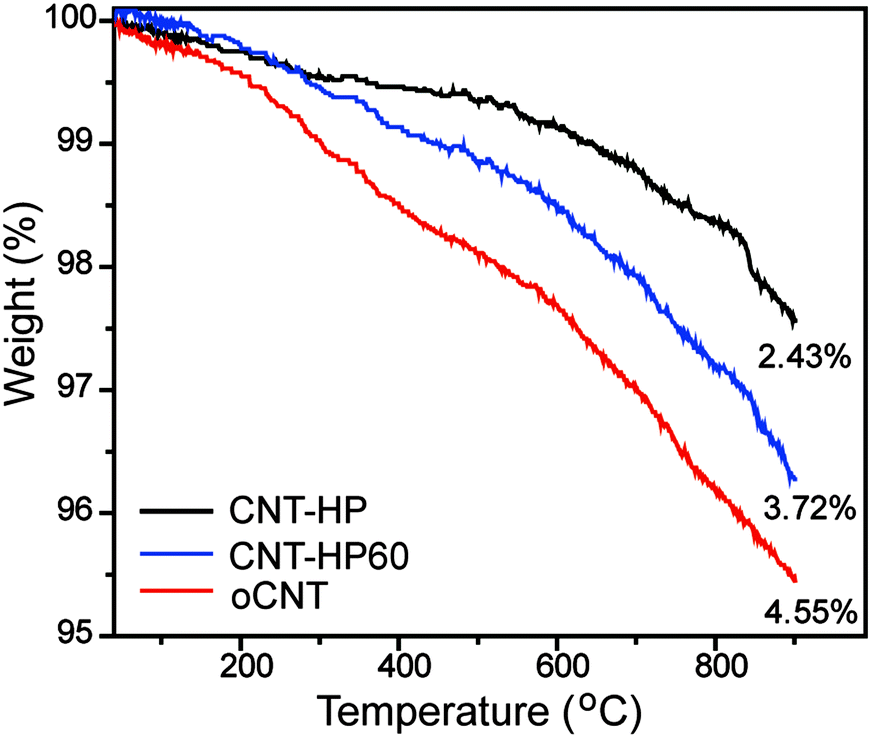 | ||
| Fig. 4 TGA curves of different CNT samples. | ||
Then we put our emphasis on CNT-HP60 and oCNTs for further study. XPS was used to analyze the surface state of the two catalysts. The binding energy was calibrated by referencing the C1s peak at 284.6 eV. The peak which was located at around 533 eV was ascribed to the O1s spectrum. Detailed information on surface oxygen functional groups would be obtained after the deconvolution of the O1s spectrum (Fig. 5). The peak labeled as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O which was centered at 531.7 eV was attributed to the carbonyl group. The peak located at 532.6 eV was ascribed to carboxylic acid, anhydride, ester and lactone, and was referred to as OC–O. The hydroxyl group as well as ether was denoted as C–O (H), and was centered at about 533.7 eV. The total contents of surface oxygen were 3.6 at% and 2.7 at% for oCNTs and CNT-HP60, respectively (Table 2). The carbonyl group on CNT-HP60 was calculated to be 0.3 at% after deconvolution, which was a little lower than that on oCNTs (0.5 at%). The contents of OC–O were 1.6 at% and 0.7 at% on oCNTs and CNT-HP60, respectively. On the other hand, there was only a small difference in the amount of C–O (H) between the two samples. We previously found that the carbonyl group, the carboxylic group and anhydride has remarkable effects on nitrobenzene reduction, while other functionalities such as the hydroxyl group, ether and lactone seemed to have no or marginal effect on the reaction. Among these groups which took great parts in the reaction, carbonyl group was highly active, while carboxylic group as well as anhydride played a significant negative role in the activity.19 As a result, a relatively less negative OC–O on CNT-HP60 may be an important reason for its higher activity.
O which was centered at 531.7 eV was attributed to the carbonyl group. The peak located at 532.6 eV was ascribed to carboxylic acid, anhydride, ester and lactone, and was referred to as OC–O. The hydroxyl group as well as ether was denoted as C–O (H), and was centered at about 533.7 eV. The total contents of surface oxygen were 3.6 at% and 2.7 at% for oCNTs and CNT-HP60, respectively (Table 2). The carbonyl group on CNT-HP60 was calculated to be 0.3 at% after deconvolution, which was a little lower than that on oCNTs (0.5 at%). The contents of OC–O were 1.6 at% and 0.7 at% on oCNTs and CNT-HP60, respectively. On the other hand, there was only a small difference in the amount of C–O (H) between the two samples. We previously found that the carbonyl group, the carboxylic group and anhydride has remarkable effects on nitrobenzene reduction, while other functionalities such as the hydroxyl group, ether and lactone seemed to have no or marginal effect on the reaction. Among these groups which took great parts in the reaction, carbonyl group was highly active, while carboxylic group as well as anhydride played a significant negative role in the activity.19 As a result, a relatively less negative OC–O on CNT-HP60 may be an important reason for its higher activity.
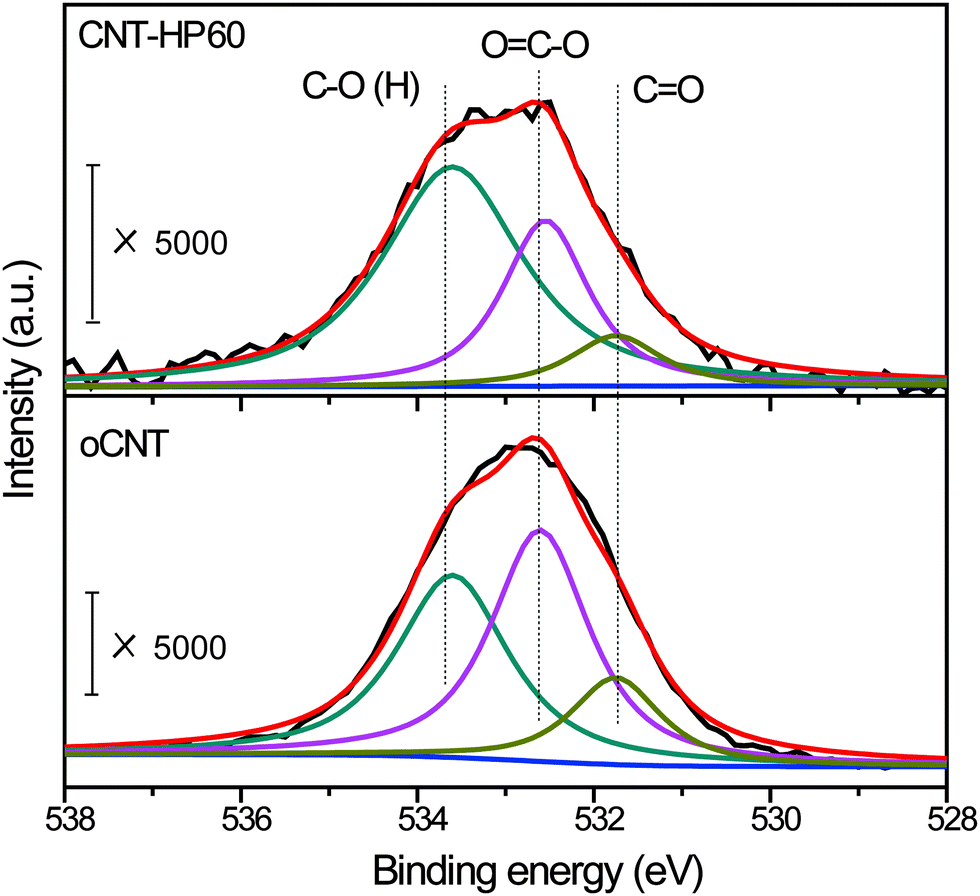 | ||
| Fig. 5 O1s XPS spectra of CNT-HP60 and oCNT. | ||
| Sample | O (at%) | CO (at%) |
OC–O (at%) |
C–O (H) (at%) |
|---|---|---|---|---|
| oCNT | 3.6 | 0.5 | 1.6 | 1.5 |
| CNT-HP60 | 2.7 | 0.3 | 0.7 | 1.7 |
The TPD analyses were performed under the same conditions for the two samples (Fig. 6). The released amounts of both CO and CO2 were higher for oCNT, suggesting that there were more oxygenated groups on oCNT. CO was derived from the decomposition of anhydride, ether, lactone, the hydroxyl group and the carbonyl group. Among these functionalities, the carbonyl group played an important role and served as a critical kind of active site, but anhydride was negative to the reaction, while the effects of other groups on the reaction could be neglected. Anhydride would decompose in the range from about 400 °C to 600 °C, but there was no remarkable difference for the CO profiles for the two samples in this range, indicating that there was no obvious difference in CO formation due to the anhydride decomposition of the two carbon catalysts. CO2 was formed by the decomposition of a carboxylic group, anhydride and lactone, all of which were negative or not reactive. It showed that more CO2 was formed from the oCNT than that from CNT-HP60 in the range from around 200 °C to 600 °C. The formation of CO2 in this range was ascribed to the decomposition of the carboxylic group and anhydride. The CO2 profiles suggested that there were more carboxylic groups and anhydrides on oCNT, which is in accordance with the XPS results. Because both functionalities played significant negative roles in nitrobenzene reduction, it was understandable that the low amount of CO2 precursors leads to the relatively high activity of CNT-HP60. Besides, there was no obvious change in functional groups on the used CNT-HP60 compared to the fresh counterpart (Table S1 and Fig. S6, ESI†).
 | ||
| Fig. 6 TPD profiles of oCNT and CNT-HP60. | ||
In summary, the CNTs could be functionalized by H2O2, and the obtained materials exhibited better catalytic performance than that for HNO3 oxidized samples in the reduction of nitrobenzene. With the assistance of several characterizations, including TGA, XPS and TPD, it suggested that the high activity of the H2O2 oxidized sample may be ascribed to the relatively low content of the negative functionalities, such as the carboxylic group and anhydride. In addition, the H2O2 oxidized sample could also effectively catalyze the reduction of some substituted nitroarenes. These findings may provide a facile method to prepare efficient carbon catalysts for green and sustainable chemistry.
This work is financially supported by MOST (2011CBA00504), NSFC of China (21133010, 51221264, and 21261160487), “Strategic Priority Research Program” of the Chinese Academy of Sciences, Grant No. XDA09030103 and the Doctoral Starting up Foundation of Liaoning Province, China (20121068). The authors thank Rui Huang and Dr Neeraj Gupta for fruitful discussions.
Notes and references
- J. S. Lee, X. Q. Wang, H. M. Luo and S. Dai, Adv. Mater., 2010, 22, 1004 CrossRef CAS PubMed.
- J. Zhang, D. S. Su, A. H. Zhang, D. Wang, R. Schlögl and C. Hébert, Angew. Chem., Int. Ed., 2007, 46, 7319 CrossRef CAS PubMed.
- J. Zhang, X. Liu, R. Blume, A. H. Zhang, R. Schlögl and D. S. Su, Science, 2008, 322, 73 CrossRef CAS PubMed.
- B. Frank, J. Zhang, R. Blume, R. Schlögl and D. S. Su, Angew. Chem., Int. Ed., 2009, 48, 6913 CrossRef CAS PubMed.
- D. R. Dreyer, H. P. Jia and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 6813 CAS.
- Y. Kuang, N. M. Islam, Y. Nabae, T. Hayakawa and M. Kakimoto, Angew. Chem., Int. Ed., 2010, 49, 436 CrossRef CAS PubMed.
- H. Huang, J. Huang, Y. M. Liu, H. Y. He, Y. Cao and K. N. Fan, Green Chem., 2012, 14, 930 RSC.
- C. L. Su, M. Acik, K. Takai, J. Lu, S. J. Hao, Y. Zheng, P. P. Wu, Q. L. Bao, T. Enoki, Y. J. Chabal and K. P. Loh, Nat. Commun., 2012, 3, 1298 CrossRef PubMed.
- X. H. Li and M. Antonietti, Angew. Chem., Int. Ed., 2013, 52, 4572 CrossRef CAS PubMed.
- H. Yu, F. Peng, J. Tan, X. W. Hu, H. J. Wang, J. Yang and W. X. Zheng, Angew. Chem., Int. Ed., 2011, 50, 3978 CrossRef CAS PubMed.
- R. S. Downing, P. J. Kunkeler and H. van. Bekkum, Catal. Today, 1997, 37, 121 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332 CrossRef CAS PubMed.
- F. Ragaini and S. Cenini, J. Mol. Catal. A: Chem., 1996, 105, 145 CrossRef CAS.
- S. Kim, E. Kim and B. M. Kim, Chem. – Asian J., 2011, 6, 1921 CrossRef CAS PubMed.
- R. V. Jagadeesh, G. Wienhöfer, F. A. Westerhaus, A. E. Surkus, M. M. Pohl, H. Junge, K. Junge and M. Beller, Chem. Commun., 2011, 47, 10972 RSC.
- B. H. Han, D. H. Shin and S. Y. Cho, Tetrahedron Lett., 1985, 26, 6233 CrossRef CAS.
- J. W. Larsen, M. Freund, K. Y. Kim, M. Sidovar and J. L. Stuart, Carbon, 2000, 38, 655 CrossRef CAS.
- Y. J. Gao, D. Ma, C. L. Wang, J. Guan and X. H. Bao, Chem. Commun., 2011, 47, 2432 RSC.
- S. C. Wu, G. D. Wen, B. W. Zhong, B. S. Zhang, X. M. Gu, N. Wang and D. S. Su, Chin. J. Catal., 2014, 35, 914 CrossRef CAS.
- S. C. Wu, G. D. Wen, J. Wang, J. F. Rong, B. N. Zong, R. Schlögl and D. S. Su, Catal. Sci. Technol., 2014, 4, 4183 CAS.
- S. C. Wu, G. D. Wen, X. M. Liu, B. W. Zhong and D. S. Su, ChemCatChem, 2014, 6, 1558 CrossRef CAS.
- S. C. Tsang, Y. K. Chen, P. J. F. Harris and M. L. H. Green, Nature, 1994, 372, 159 CrossRef CAS.
- J. Zhang, H. L. Zou, Q. Qing, Y. L. Yang, Q. W. Li, Z. F. Liu, X. Y. Guo and Z. L. Du, J. Phys. Chem. B, 2003, 107, 3712 CrossRef CAS.
- I. D. Rosca, F. Watari, M. Uo and T. Akasaka, Carbon, 2005, 43, 3124 CrossRef CAS PubMed.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833 CrossRef CAS PubMed.
- Y. Miyata, Y. Maniwa and H. Kataura, J. Phys. Chem. B, 2006, 110, 25 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental section, catalytic performance of two CNT based samples, repeated tests, nitrogen adsorption isotherms and TPD profiles of the fresh and used catalysts. See DOI: 10.1039/c4cp04658g |
| This journal is © the Owner Societies 2015 |