
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ab initio multiple cloning simulations of pyrrole photodissociation: TKER spectra and velocity map imaging†
Dmitry V.
Makhov
a,
Kenichiro
Saita‡
a,
Todd J.
Martinez
bc and
Dmitrii V.
Shalashilin
a
aDepartment of Chemistry, University of Leeds LS2 9JT, UK. E-mail: D.Makhov@leeds.ac.uk; D.Shalashilin@leeds.ac.uk
bDepartment of Chemistry and The PULSE Institute, Stanford University, Stanford, CA 94305, USA. E-mail: toddjmartinez@gmail.com
cSLAC National Accelerator Laboratory, Menlo Park, CA 94025, USA
First published on 11th December 2014
Abstract
We report a detailed computational simulation of the photodissociation of pyrrole using the ab initio Multiple Cloning (AIMC) method implemented within MOLPRO. The efficiency of the AIMC implementation, employing train basis sets, linear approximation for matrix elements, and Ehrenfest configuration cloning, allows us to accumulate significant statistics. We calculate and analyze the total kinetic energy release (TKER) spectrum and Velocity Map Imaging (VMI) of pyrrole and compare the results directly with experimental measurements. Both the TKER spectrum and the structure of the velocity map image (VMI) are well reproduced. Previously, it has been assumed that the isotropic component of the VMI arises from long time statistical dissociation. Instead, our simulations suggest that ultrafast dynamics contributes significantly to both low and high energy portions of the TKER spectrum.
I. Introduction
The dynamics of electronically nonadiabatic reactions is key to understanding many processes in chemistry and biochemistry. For example light harvesting in plants, fluorescence of living organisms, and visual reception all involve photochemical reactions that include electronic excitation and subsequent electronically nonadiabatic dynamics. Recently significant progress has been made in experimental ultrafast time resolved spectroscopy studies of various photochemical reactions, focused on biologically related molecules. The derivatives of heteroaromatic molecules such as pyrrole, imidazole, and phenol are important chromophores of nucleobases and aromatic amino acids. The mechanisms of their photochemistry have been a focus of experimental1 and theoretical2 attention. It was suggested that the N–H/O–H bond fission was an important channel in the photodissociation dynamics and the role of the 1πσ* states in this process has been emphasized.3 Total kinetic energy release (TKER) spectra and velocity map images (VMI) for various bond fission reactions were reported providing invaluable information but to the best of our knowledge VMI and TKER spectra have never been calculated theoretically for these molecules. This is a difficult task because TKER spectra reflect important details of quantum dynamics in multidimensional systems, where realistic calculations beyond simple reduced dimensionality models are challenging.In particular, the photodissociation of pyrrole (C4NH5), prototypical of many heteroaromatic systems, has been investigated.1a,4 N–H bond fission is the primary decay mechanism for pyrrole molecules excited to the πσ* state. Dissociating H atoms have been detected5 with a centre-of-mass frame energy of 18 kcal mol−1 and a strong anisotropy, indicating prompt dissociation (within the molecular rotation period). These initial findings were supported by further evidence from ion-imaging1b and the hydrogen (Rydberg) atom photofragment translational spectroscopy (HRAPTS)4a experiments. The high resolution HRAPTS data showed several notable features, including direct (Herzberg–Teller allowed) excitation into the 1A2 electronic state, with preservation of the H–T active vibrations into the product radical. Time resolved studies have also been performed6,7 albeit in the region of the stronger1 absorption.
Previous quantum dynamics calculations1b,2c,d have been performed using reduced dimensionality model PESs. These have predicted rapid dissociation and significant population of the 2B1 state of pyrrolyl, which is difficult to reconcile with the experimental data. Theoretical work by Sobolewski and Domcke2a,b showed the existence of conical intersections in the decay channel, which are by now well-known and opened the possibility of electronic population branching during the dissociation. Despite the fact that both 2A2 and 2B1 states of pyrrolyl are energetically accessible, the interpretation of the HRAPTS data by experimentalists is that population was only observed in the lowest (X2A2) electronic state of pyrrolyl. The peaks in the TKER spectrum were accordingly assigned to various vibrational states of this ground electronic state of pyrrolyl radical.
Recently we performed our first simulation of the dynamics of photodissociation of pyrrole8 using the Multiconfigurational Ehrenfest (MCE)9 method. MCE uses Ehrenfest trajectories dressed with Gaussian wave packets, which serve as a basis set for quantum propagation of the nuclear wave packet. Each of these trajectory basis functions (TBFs) has an associated complex time-dependent amplitude and they are all coupled to each other (through the nuclear Schrodinger equation). Thus, the MCE method is fully quantum mechanical in principle. However, the underlying equations of motion in the Ehrenfest method can be problematic in regions where electronic states are decoupled. Detailed balance is not obeyed10 and the potential energy surfaces governing the propagation are averages of the adiabatic states.11 With a sufficiently large basis set, these problems would be corrected by the solution of the time-dependent Schrodinger equation for the amplitudes of the basis functions. However, this could require an impractical number of basis functions. We recently showed12 how these problems can be overcome by introducing cloning of Ehrenfest configurations, analogous to the spawning procedure for adaptive basis set expansion used in ab initio multiple spawning (AIMS).13
The ab initio multiple cloning (AIMC) method is based on MCE dynamics including cloning and coupled to an electronic structure package to calculate the potential energy surfaces, gradients, and nonadiabatic coupling matrix elements “on the fly” as needed by the dynamics. The dynamics calculations are carried out in full dimensionality, i.e. all degrees of freedom are included explicitly. Similarly to AIMS, the AIMC method uses a basis of trajectory guided Gaussian wave packets to represent the motion of the nuclei.
Although AIMC differs from AIMS in the equations of motion for the TBFs, the methods are nevertheless similar enough to be implemented within the same code, and both methods have been implemented in MolPro. Recently we developed a new implementation of the MCE method within the AIMS code, which combines the best features of the two methods. Our ab initio Multiple Cloning (AIMC) approach12 has a number of useful features:
(1) The trajectory basis functions are propagated using Ehrenfest equations of motion. These can be quite accurate in regions where two or more electronic states are strongly coupled and may then be preferable to classical equations of motion on a specific electronic state.
(2) As discussed above, the Ehrenfest equations of motion can be problematic after passing a strong coupling region. There Ehrenfest trajectories, which are guided by potential energy surface average of those of individual electronic states, may move outside of the dynamically important region. This is remedied in the AIMC method by a procedure called cloning, which is an adaptation of spawning procedure of AIMS. Cloning reprojects the Ehrenfest trajectories on the individual electronic states after quantum transitions are completed. Importantly, this projection is done in a manner that does not alter the nuclear wavefunction at the time of cloning (similar to spawning in AIMS). The AIMC method combines the advantages of the AIMS and MCE methods.
(3) AIMC uses an interpolation procedure for calculating matrix elements that is based solely on quantities calculated at the centers of the TBFs. There are no calculations of energies, gradients, or nonadiabatic coupling matrix elements at geometries intermediate between trajectory centers. This minimizes the number of expensive ab initio electronic structure calculations required during the dynamics. Additionally, it is possible to calculate many of the underlying Ehrenfest trajectories independently and then to later recombine them in a “post-processing” procedure. The quantum amplitudes are calculated in this post-processing stage. We refer to this as “incremental propagation” below and it has the advantage that quantum mechanical coupling can be computed at almost no cost in comparison to the ab initio “on the fly” calculation of the ensemble of Ehrenfest configuration TBFs.
(4) AIMC makes use of the idea of train basis sets14 (also known as time-displaced basis functions15). Train basis functions follow each other along the same Ehrenfest trajectory but with a time delay, so that propagating trains does not require additional electronic structure ab initio information. Train basis sets reduce the noise and improve the quality of quantum dynamics calculations at almost no extra cost.
Like other ab initio methods, such as Multiple Spawning13b–e and Variational Gaussians,16 AIMC simulates the dynamics in full dimensionality and without precomputed potential energy surfaces. Potential energy surfaces and their coupling matrix elements are calculated “on the fly” by solving the electronic Schrodinger equation. The selection of a few important degrees of freedom for the dynamics is avoided, in some cases allowing one to see that unexpected coordinates are important. AIMC (and also AIMS and previous ab initio MCE simulations) are not biased by the choice of which coordinates to model, since all are included on equal footing. The ab initio MCE method (without cloning) has been used previously for a more complete analysis of the dissociation of pyrrole.8 This showed that the 2B1 state of the pyrrolyl radical is only weakly populated, confirming the original assignment of the HRAPTS data by experimentalists. We noticed that after partial departure of the H-atom, the two electronic states 2A2 and 2B1 are still coupled via an intersection between the potential energy surfaces of the pyrrolyl radical, which is nearly isolated. This intersection is reached by the pyrrolyl vibrational motion, and as a result the population of the higher 2B1 state resulting from rapid nonadiabatic dynamics of the N–H bond fission leaks to the lower 2A2 state via longer time dynamics at this final intersection. A possible role of this feature has been proposed previously,2c but a multidimensional simulation shows it clearly. Previously, such a conical intersection between two lowest electronic states of pyrrolyl has been found17 and our current simulation suggests that it may play an important role in the nonadiabatic dynamics of pyrrole.
In this paper we report a more detailed simulation of pyrrole dissociation after excitation to the first excited state, which corresponds precisely to the conditions of the recent experiment1a where the TKER spectrum was measured after 250 nm excitation of pyrrole. The experiment1a uses a 120 fs pump pulse, creating a wave packet in the Franck–Condon region, which is conveniently modeled with AIMC. The experiment also exploits the fact that the transition dipole moment for the chosen excitation can be well approximated as perpendicular to the N–H bond in pyrrole. Thus, the angular distribution of dissociated H atoms with respect to the pump pulse (velocity map imaging or VMI) provides information about the detailed reaction dynamics. Our AIMC calculation reproduces very well both the TKER spectrum and the strong anisotropy of the VMI observed in recent experiments,1a validating the mechanistic information inferred from the simulations.
II. Theory
II.1 Working equations
We start with a brief sketch of the MCE method and its implementation, as full details have been provided previously.12 In the Multiconfigurational Ehrenfest (MCE) approach, the wave-function is represented in a basis set of TBFs |ψn(t)〉 composed of nuclear and electronic parts: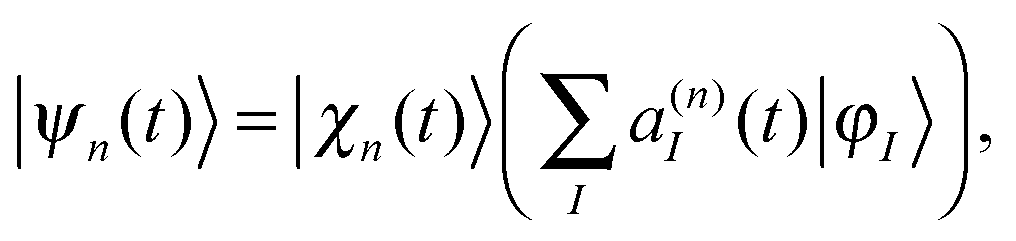 | (1) |
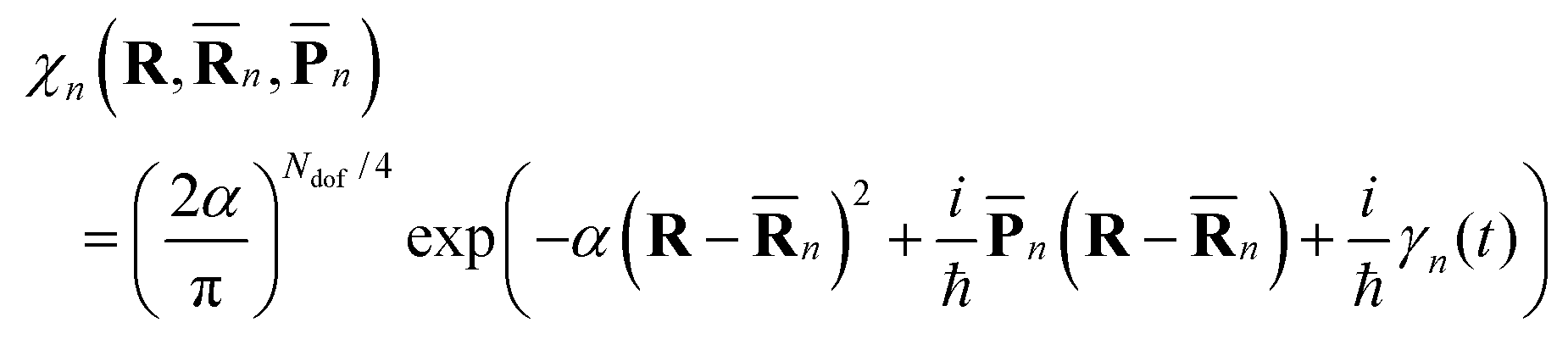 | (2) |
![[R with combining macron]](https://www.rsc.org/images/entities/b_char_0052_0304.gif) n(t) and
n(t) and ![[P with combining macron]](https://www.rsc.org/images/entities/b_char_0050_0304.gif) n(t) are the phase space coordinate and momentum vectors of the n-th basis function center, and γn is a phase which evolves as:
n(t) are the phase space coordinate and momentum vectors of the n-th basis function center, and γn is a phase which evolves as: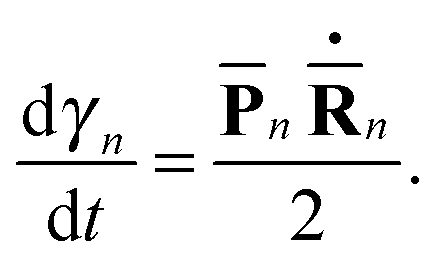 | (3) |
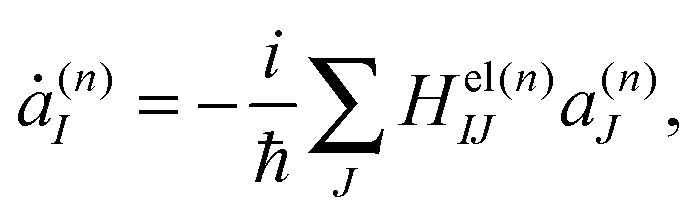 | (4) |
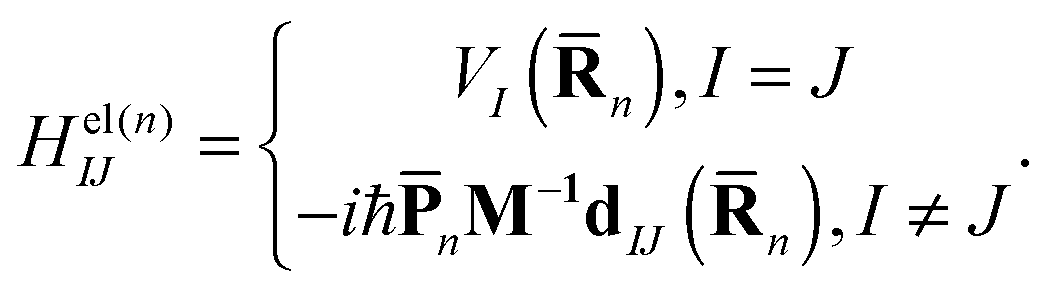 | (5) |
n) is the Ith potential energy surface and dIJ(n) = 〈φI|∇R|φJ〉 is the Nonadiabatic Coupling Matrix Element (NACME), and M is the diagonal matrix of nuclear masses.
The centers of the Gaussian basis functions follow Newton’s equations of motion:
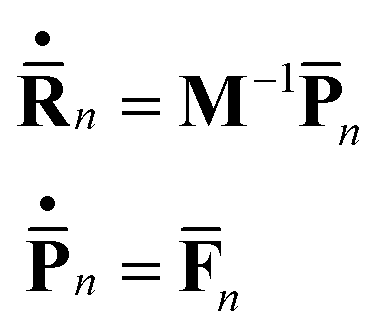 | (6) |
![[F with combining macron]](https://www.rsc.org/images/entities/b_char_0046_0304.gif) n includes both the usual gradient term and also the Hellmann–Feynman force20 related to nonadiabatic coupling:
n includes both the usual gradient term and also the Hellmann–Feynman force20 related to nonadiabatic coupling: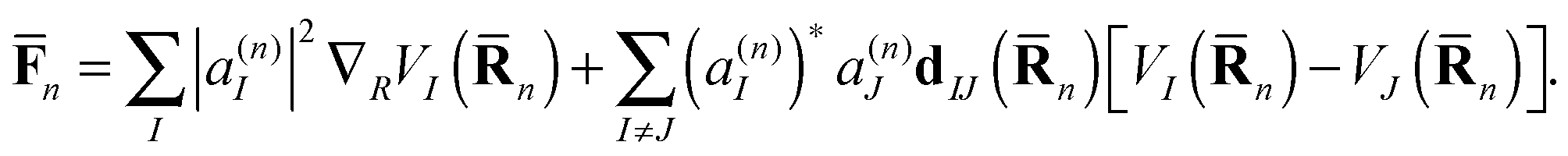 | (7) |
A single Ehrenfest configuration is not flexible enough to accurately describe quantum dynamics. In the MCE approach, multiple Ehrenfest configurations are used and these represent a basis set in which the total wavefunction is expanded as:
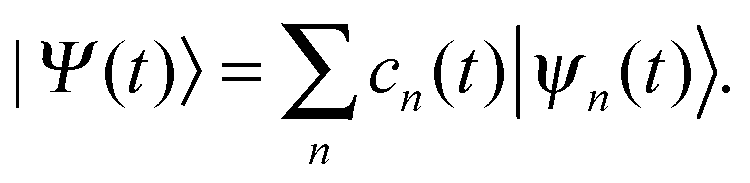 | (8) |
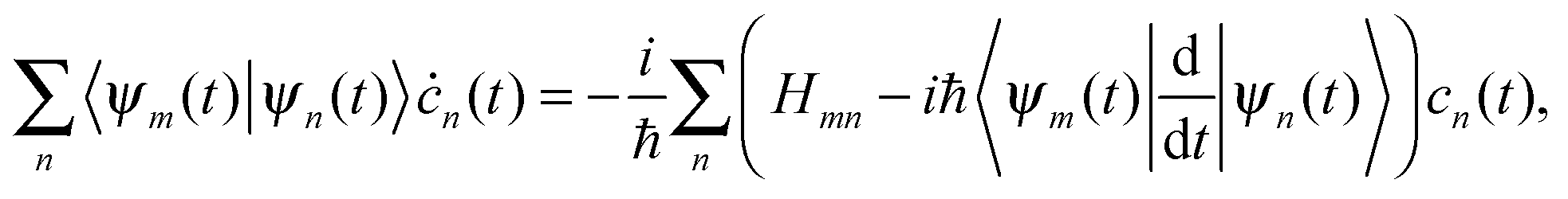 | (9) |
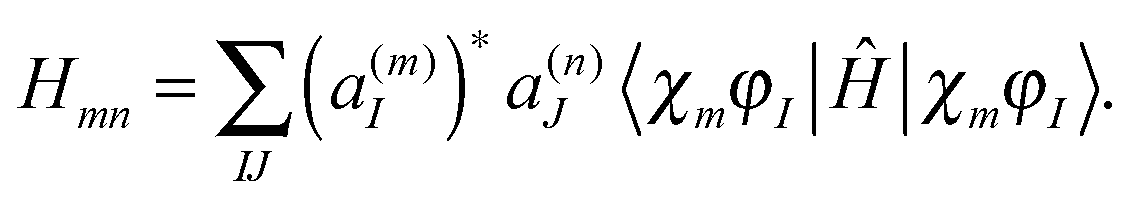 | (10) |
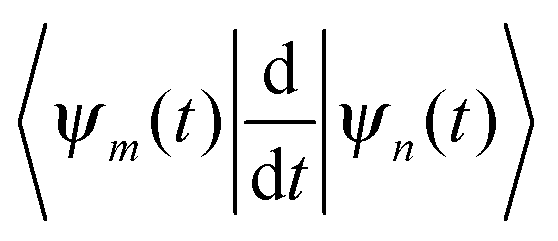 originates from time dependence of the TBFs and is given by:
originates from time dependence of the TBFs and is given by: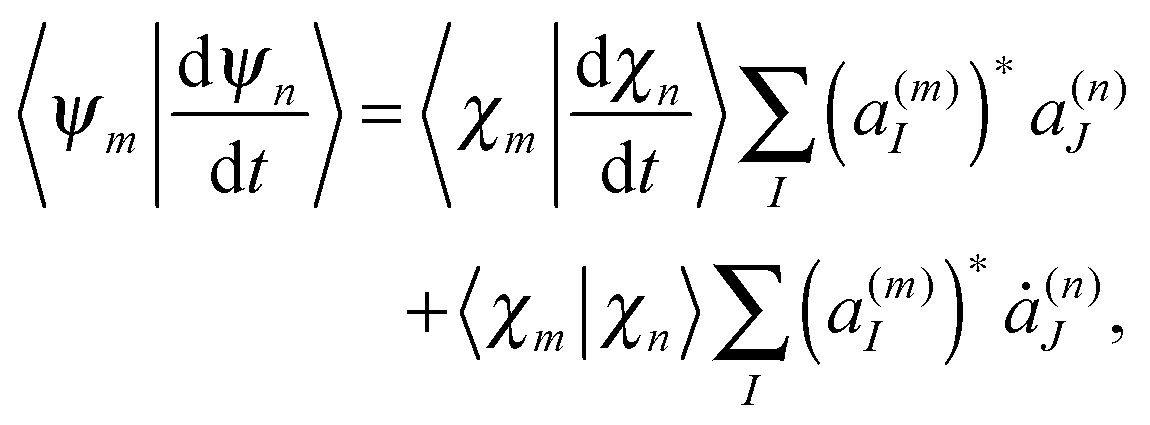 | (11) |
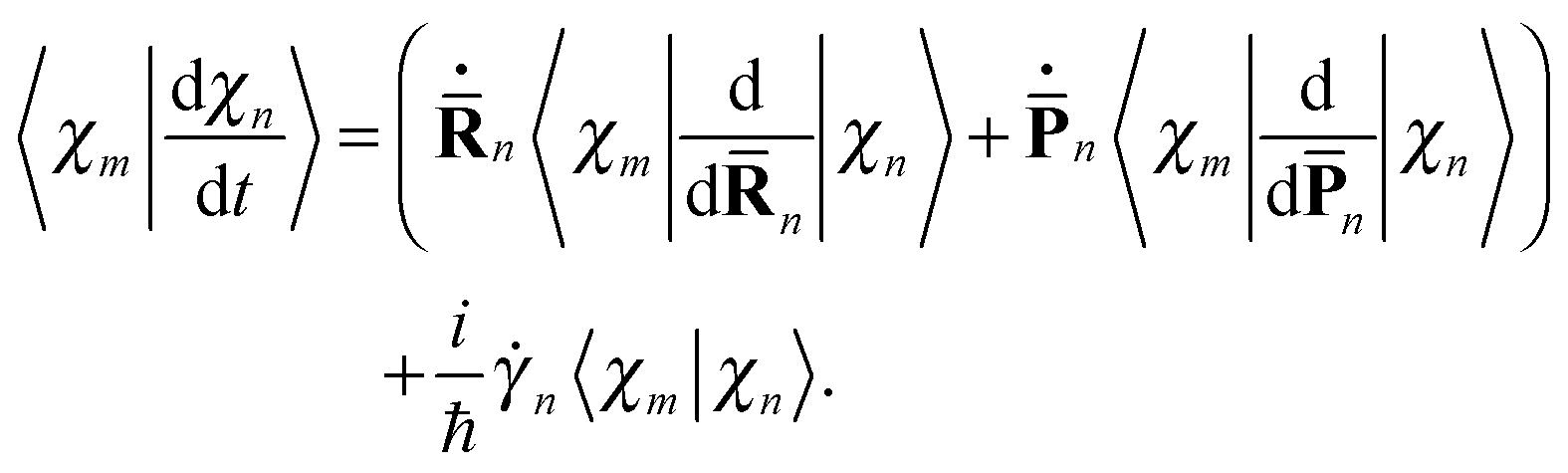 | (12) |
II.2 Matrix element approximations
Assuming that the electronic wave function depends weakly on R, so that the second derivative with respect to R can be disregarded, the matrix elements of the Hamiltonian can be written as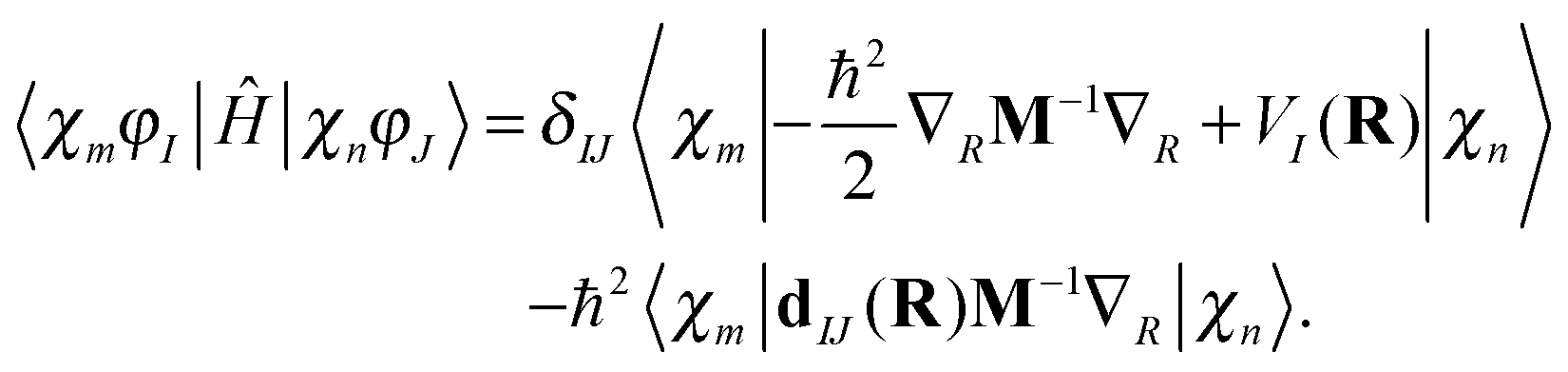 | (13) |
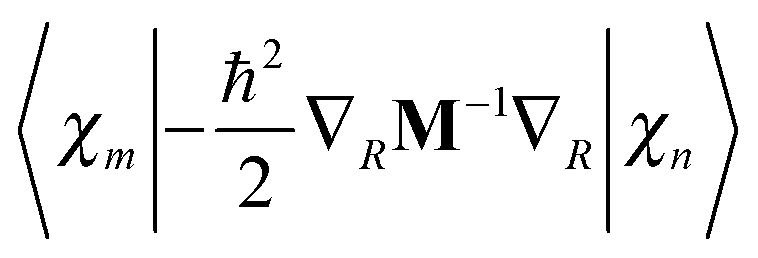 is easily calculated analytically. MCE makes some very simple but reasonable approximations for the matrix elements of the potential energy and nonadiabatic coupling, as described previously:12
is easily calculated analytically. MCE makes some very simple but reasonable approximations for the matrix elements of the potential energy and nonadiabatic coupling, as described previously:12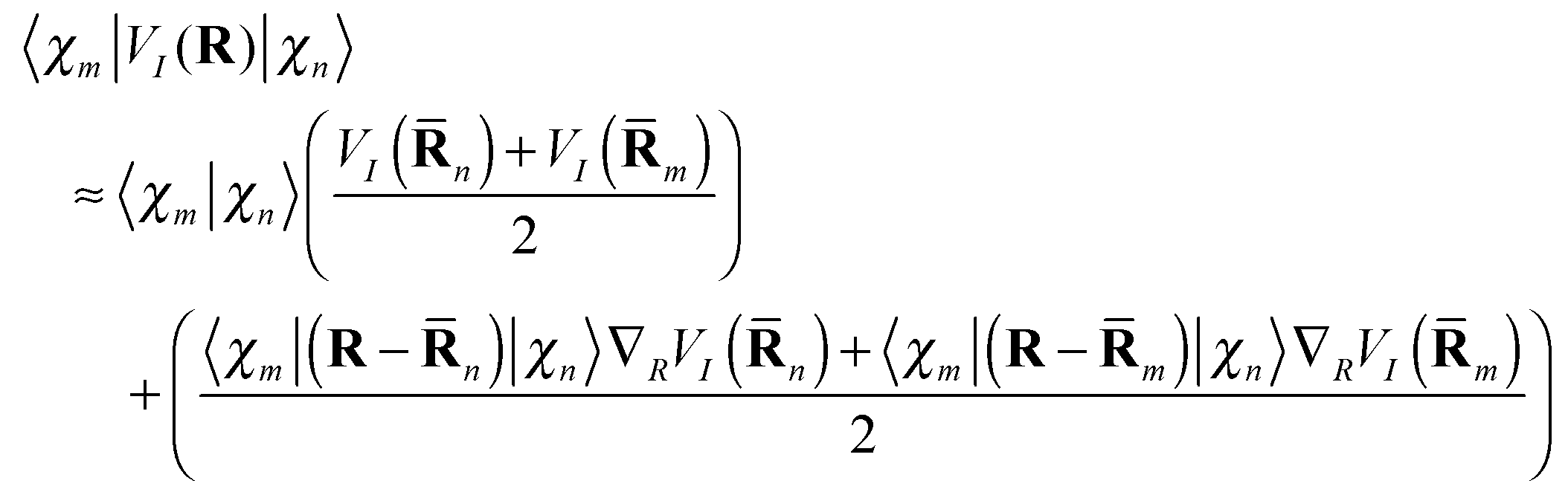 | (14) |
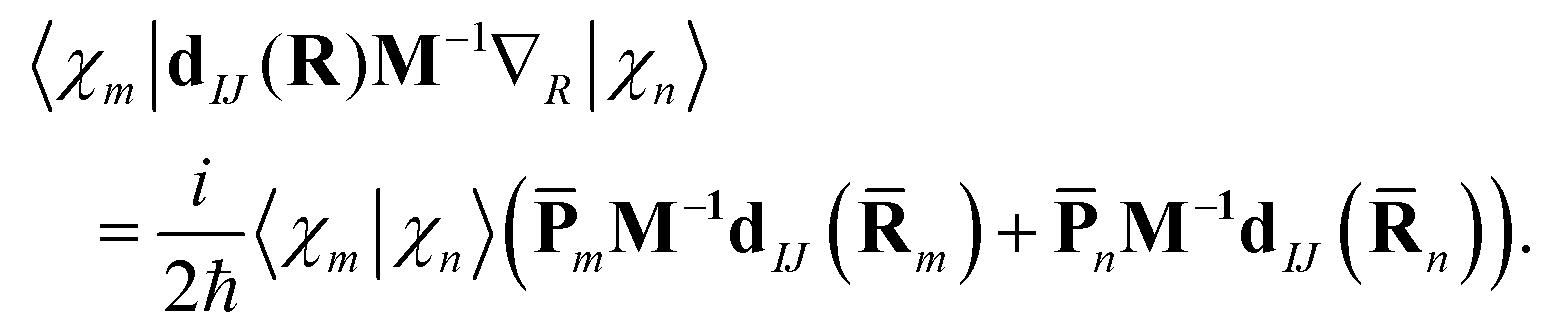 | (15) |
II.3 Basis sets: cloning, trains, and incremental propagation
The Ehrenfest basis set is guided by an average potential which can be advantageous when quantum nonadiabatic coupling is strong and quantum transitions are frequent, but it also becomes unphysical when two or more electronic states have significant amplitudes and very different shape (leading to wave packet branching after leaving the nonadiabatic coupling region).In order to reproduce the branching of the wave function, we adopted the spawning procedure from MS method into the so called Ehrenfest configuration cloning,12 where a basis function is replaced by two functions, each guided by a single potential energy surface. After cloning, an Ehrenfest configuration 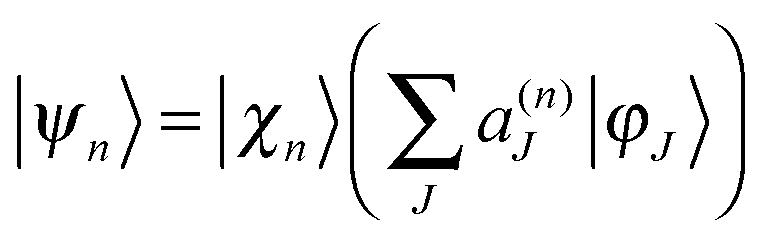 yields two configurations:
yields two configurations:
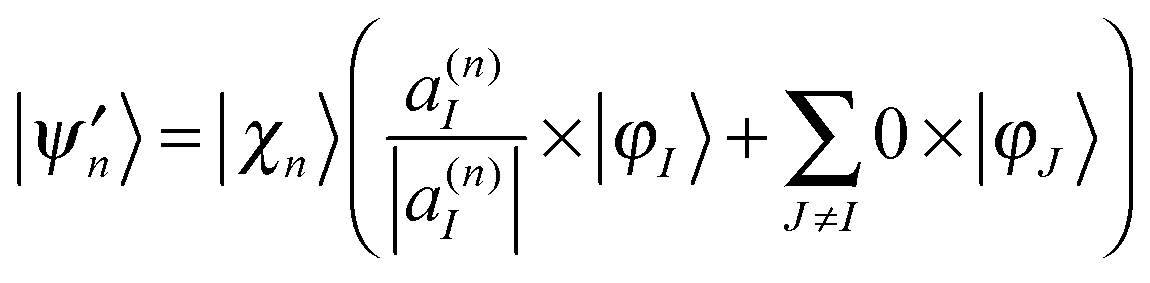 | (16) |
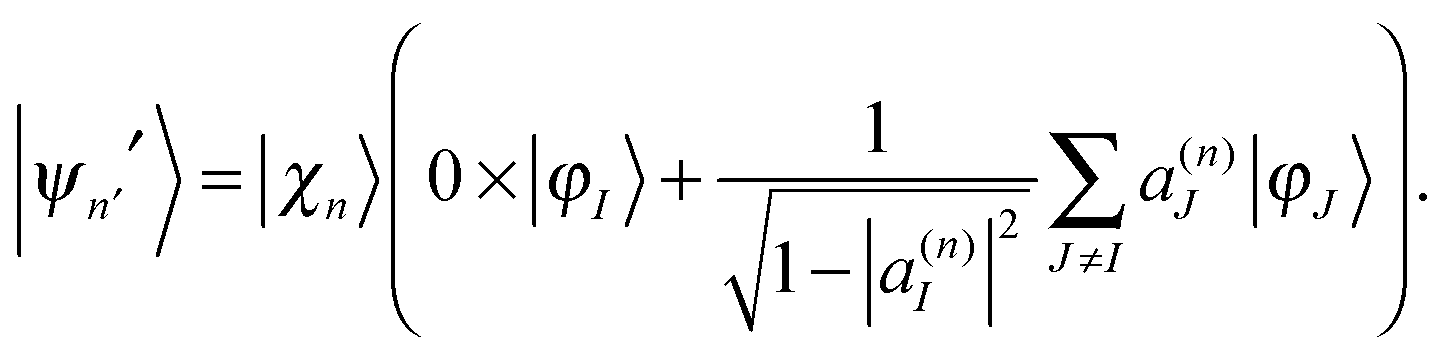 | (17) |
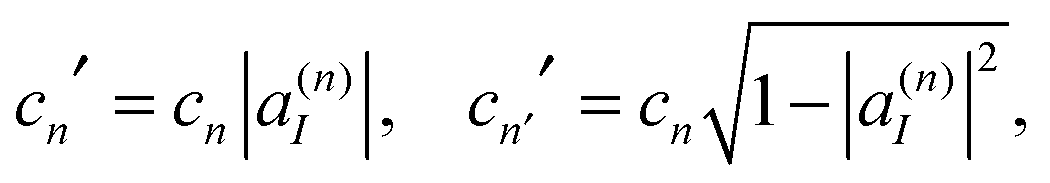 | (18) |
| cn|ψn〉 = cn′|ψn′〉 + cn′′|ψn′′〉. | (19) |
We define a “breaking force” for each state in an Ehrenfest TBF:
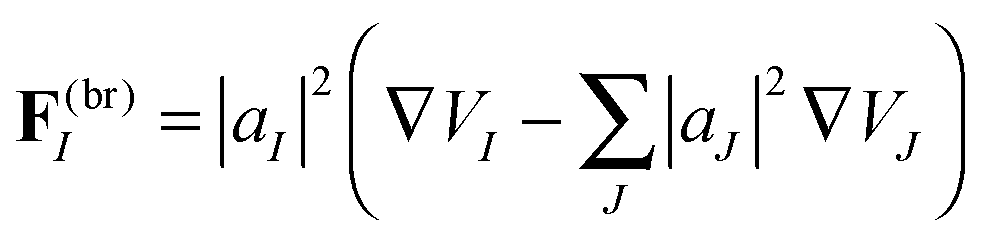 | (20) |
The solution of the Schrodinger eqn (9) was performed using coherent state train basis sets,13a,14 where Gaussian basis functions are moving along the same Ehrenfest trajectory but with a time-shift Δt. This approach ensures the preservation of the interaction between basis-functions during the run, and the same electronic structure data are used repeatedly by all basis-functions in the train drastically reducing the computational cost.
We also have used the incremental propagation, also referred to as “bit-by-bit” propagation.9c In this procedure, the initial wavefunction is expanded as a superposition of Gaussian wavepackets. Subsets of these Gaussian basis functions (the “bits”) are then propagated independently. First, each “bit” gives an origin to a trajectory, which can branch by cloning. Then, each trajectory defines a single train basis for propagating the eqn (9). After all trajectories describing the initial “bits” of the wavefunction have been calculated with the help of eqn (4) (including cloning) and the basis trains have been set, a post-processing step solves the eqn (9) for the coupled amplitudes of the of train basis functions in each “bit”. The matrix element approximations discussed in Section II.2 make it efficient and convenient to do this as a post-processing step because all of the needed electronic structure information can be stored and does not need to be recalculated. Finally, the contribution of the “bits” is properly averaged. Although the “bits” are not coupled with each other, incremental “bit-by-bit” propagation is not an approximation but rather a convenient way to improve sampling, replacing propagation of the whole wave function with a large basis by many smaller basis set propagations. “Bit-by-bit” approach is accurate provided that each “bit” is propagated exactly, which is true on a short time scale. Fig. 1 sketches all sampling ideas used in the AIMC approach. The bits of the initial wave function shown by blue circles generate basis set trains, which are propagated and cloned after passing the intersections.
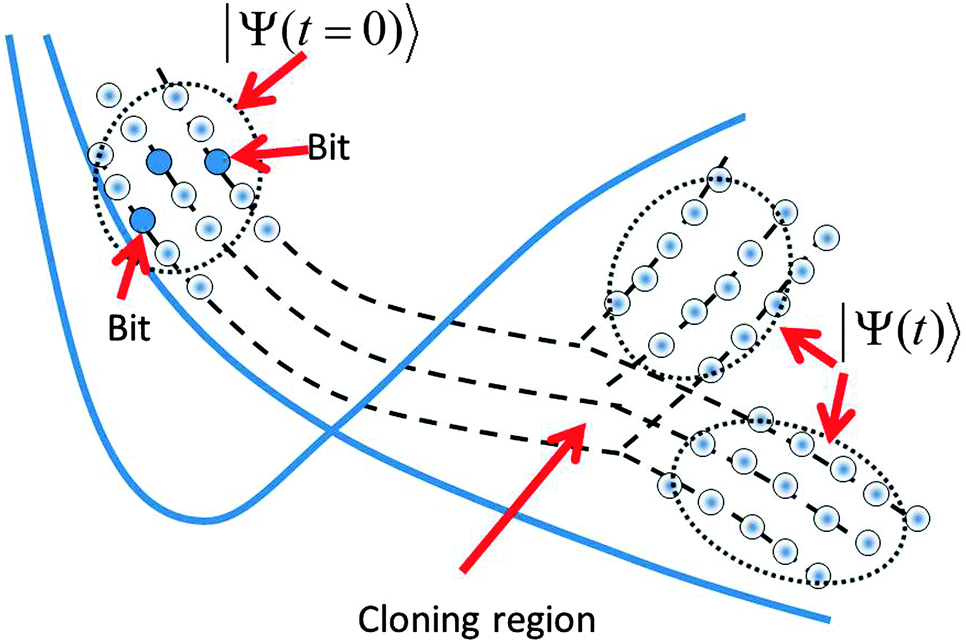 | ||
| Fig. 1 A sketch of the AIMC propagation scheme. First the wave function is represented as a superposition of Gaussian Coherent States (“bits”), each of which is a central trajectory for a train basis set. After passing the intersection the trains branch in the process of cloning (a similar diagram would apply to MCE, without the cloning step). | ||
Apart from trivial differences in the notations, AIMC12 differs from ab initio MCE8,9 in the introduction of cloning, time displaced basis sets or trains, and a more accurate estimation (14) of the matrix element of the potential energy. On the other hand in the ref. 8 and 9 more than one trajectory was originated from each “bit” providing the basis set for the propagation of eqn (9). In this work only one trajectory per “bit” was used to provide a train basis set but in principle the use of more than one train per “bit” with coupling between the trains is possible, although at higher computational cost.
III. Computational details
In previous TKER experiments on pyrrole,3a,4a,19 a long resonance pulse was used, creating a narrow band of electronically excited vibrational states. In contrast, the most recent experiment1a uses a short femtosecond pulse and creates a Franck–Condon vibrational wave packet, which is broad in energy but localized in phase space. As these initial conditions are closely related to the TBFs in the AIMC approach, this femtosecond experiment is well suited for simulation with AIMC.Using our AIMC approach, we simulated the dynamics of pyrrole following excitation to the first excited state. Trajectories were calculated using AIMS-MOLPRO,14 which was modified to incorporate Ehrenfest dynamics. Electronic structure calculations were performed with the complete active space self-consistent field (CASSCF) method using the cc-pVDZ basis set. As in previous work,8 we used an active space of eight electrons in seven orbitals (three ring π orbitals and two corresponding π* orbitals, one σ orbital and a corresponding σ* orbital). State averaging was performed over four singlet states using equal weights, i.e. the electronic wavefunction is SA4-CAS(8,7)/cc-pVDZ. The width of Gaussian functions α was taken as 4.7 Bohr−2 for hydrogen, 22.7 Bohr−2 for carbon, and 19.0 Bohr−2 for nitrogen atoms, as suggested previously.18 Three electronic states were taken into consideration during the dynamics – the ground state and the two lowest singlet excited states.
Calculations were run for maximum 200 fs with time-step ∼0.06 fs (2.5 a.u.). CASSCF calculations for pyrrole are extremely expensive, and each trajectory requires around a month of CPU time. In order to reduce computational time, calculations for the majority of trajectories were stopped as soon as N–H distance exceeded 3 Å, excluding a small number of trajectories for which simulations exhibiting N–H dissociation were carried out to a full 200 fs in order to investigate the post-dissociation dynamics of pyrrolyl radical. Although this limitation makes impossible proper adiabatic state population analysis, it does not affect TKER spectrum as the interaction between departed H-atom and the radical is extremely small at 3 Å distance.
The initial Ehrenfest configurations were randomly sampled from the ground state vibrational Wigner distribution in the harmonic approximation. The transition from the ground to the first excited state is symmetry-forbidden in the Franck–Condon approximation and only occurs due to the coordinate-dependence of the transition dipole moment. In this work, we approximate the photoexcitation by simply lifting the ground state wavepacket to the excited state, as would be appropriate for an instantaneous excitation pulse within the Condon approximation. Of course, the fine details of the initial photoexcited wavepacket are not completely accounted for in this approximation (which assumes the transition dipole moment for the transition is finite and independent of nuclear coordinates). We do not expect these details to have much effect on the observables shown in this paper, but future work can verify this by including the coordinate dependence of the transition dipole moment in the description of the excited state wavepacket.
Another interesting feature of the experiment1a is that it largely relies on the fact that only the components of the field which are perpendicular to the N–H bond can excite the A2 electronic state of pyrrole. See ESI† for more details.
In these AIMC simulations, we have used 1000 initial “bits” with one initial Ehrenfest configurations per bit. These 1000 initial “bits” (and all the configurations they clone) were recombined according to the incremental propagation described above. Cloning was applied to TBFs when the breaking acceleration of eqn (20) exceeded a threshold of 5 × 10−6 a.u. and the norm of the non-adiabatic coupling vector was simultaneously less than 2 × 10−3 a.u. After the cloning event, each branch was ascribed a proper weight which was applied in the statistical analysis of the results. A total of 91 cloning events occurred, leading to 1091 TBFs. Out of this number, 452 trajectories led to the dissociation of pyrrole molecule; the majority of the remaining 639 trajectories did not reach an intersection within the 200 fs simulation time. Cloning contributed significantly to the number of dissociating trajectories and their final distribution. We performed the full AIMC calculation, which included the solution of eqn (9) for long and bifurcating trajectories using the trains made of N = 21 Gaussians, separated by 10 time steps. If only one Ehrenfest trajectory per bit (and therefore only one train per bit) is used then the coupling between the basis Ehrenfest configurations within the train does not affect the TKER spectrum. The coupling between the amplitudes c of Ehrenfest configurations does not contribute everywhere except the cloning region, where it can result in some changes in the relative amplitudes and statistical weight of “clones”. Thus, the coupling described by eqn (9) is not very significant on the occasion of calculating TKER spectrum but still was taken into account.
IV. Results
In the analysis that follows we take into account the weights of branches wn, the sum of which is one for each initial trajectory. Fig. 2 shows the distribution of the dissociation time, which is defined as the earliest time when the N–H distance exceeds 2.5 Å. It can be seen that 35% of trajectories dissociate within 200 fs, and most of these (27% of the whole) dissociate within the first 50 fs after photoexcitation. We did not find any striking difference in the dynamics after passing the intersection between the trajectories that dissociate within 50 fs and those that dissociate in the 50–200 fs window.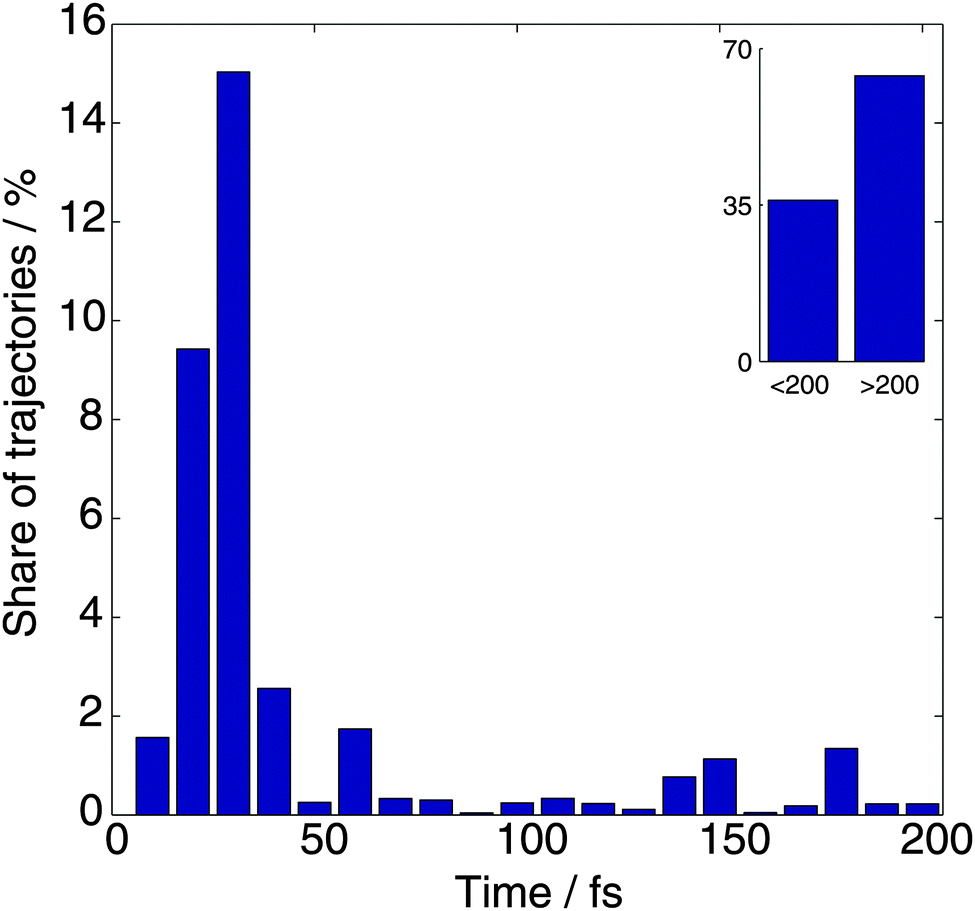 | ||
| Fig. 2 The distribution of dissociation times for the TBFs, which is defined as the first time when the N–H distance exceeds 2.5 Å. | ||
The kinetic energy distribution of the ejected hydrogen atom is presented in Fig. 3 together with the experimental observations of Stavros et al.1a Both distributions clearly exhibit two contributions: a large peak at higher energies, and a small contribution at lower energies. While the calculated kinetic energies are on average about 1.5 times higher than experimental values, this difference can be ascribed to a limited accuracy of CASSCF potential energy surfaces. To evaluate the error introduced by CASSCF PES calculations, we performed multireference perturbation theory calculations with multistate corrections (MS-CASPT2) at the Franck–Condon (FC) region of the S1 PES and the asymptotic (N–H distance of 3.0 Å) region of the S0 state and noticed that CASSCF overestimates by 1855 cm−1 the difference between the zero point energy corrected potential energies in these two regions. This means that for a more accurate MS-CASPT2 PES the kinetic energy peak would shift by approximately ∼1800–1900 cm−1 towards the lower energies improving significantly the agreement with experiment.
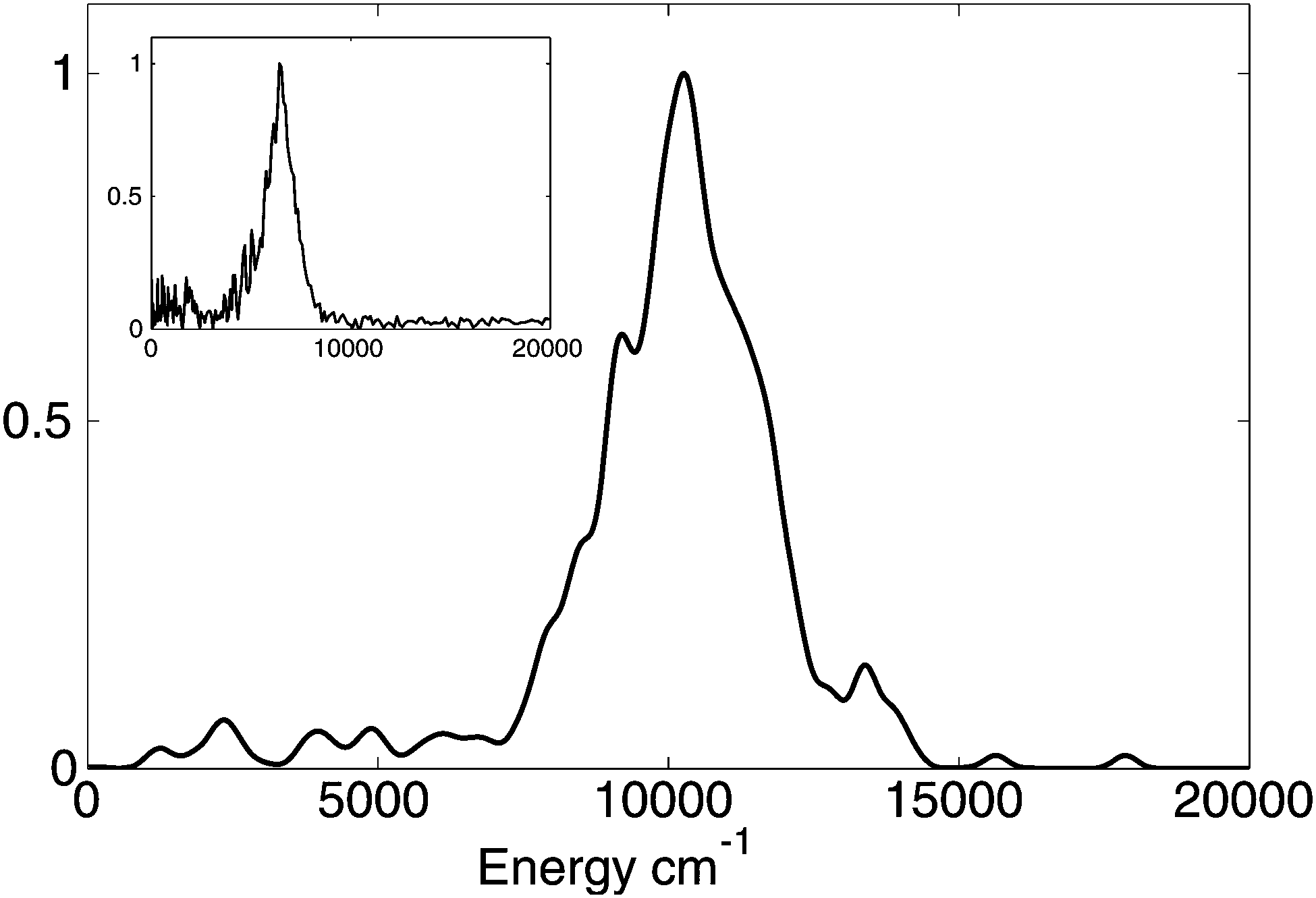 | ||
| Fig. 3 Calculated probability distribution of the kinetic energy of hydrogen atom after the dissociation. The curve is smoothed by replacing delta-functions with Gaussian functions (σ = 200 cm−1). The inset shows the experimentally measured distribution.1a | ||
The TKER spectrum alone cannot distinguish products in the ground electronic state with high vibrational energy from products in a higher electronic state with low vibrational energy. However, it is easy to make this distinction from our AIMC simulations. Analysis of the electronic state amplitudes in the Ehrenfest configurations (1) shows that the bifurcation of trajectories while passing through a conical intersection plays an important role in the formation of a two-peak spectrum: high kinetic energy product is predominantly in the ground state, confirming the conclusion of our previous work, while the low energy peak is formed by mostly low-weight branches with substantial contribution from excited electronic states (see the examples of trajectories below). This suggests that MCE method (without cloning) would not be able to reproduce correctly the character of TKER spectrum. Performing additional MCE calculations for pyrrole in order to compare the spectra directly would be too expensive, but our calculations for imidazole, which will be published later, support this suggestion: MCE method indeed gives a TKER spectrum with just one peak, while AIMC calculations correctly reproduce two-peak character of the spectrum. More detailed information is available from the angular distribution of hydrogen atoms with respect to the direction of the field. Reproducing this velocity map image represents a challenge for theory, as it is a more sensitive indicator of the dynamics than the TKER spectrum.
Fig. 4 depicts the velocity distribution of hydrogen atoms with respect to the orientation of the molecule; parts (A) and (B) show velocities in the x-direction vs. their projections on the yz plane and in the y-direction vs. xz-projection, respectively. It can be seen that high-energy hydrogen atoms travel primarily in the direction of the N–H bond, while the velocity vectors of low-energy atoms, although still lying near the xz-plane, are distributed more or less isotropically in this plane. Comparing these results with experimental data1a that high-energy atoms are predominantly ejected perpendicular to the laser polarization and the ejection of low-energy atoms is isotropic, we must conclude that transition dipole moment is orthogonal to the N–H bond for the excitations leading to high-energy ejections, and lies in the xz-plane for the ones leading to low-energy ejections. Both conditions are satisfied if the transition dipole is pointed in x-direction, i.e. orthogonal to the molecule plane. However, we cannot rule out that different mechanisms (with different orientations of transition dipoles) can be responsible for high- and low-energy ejections.
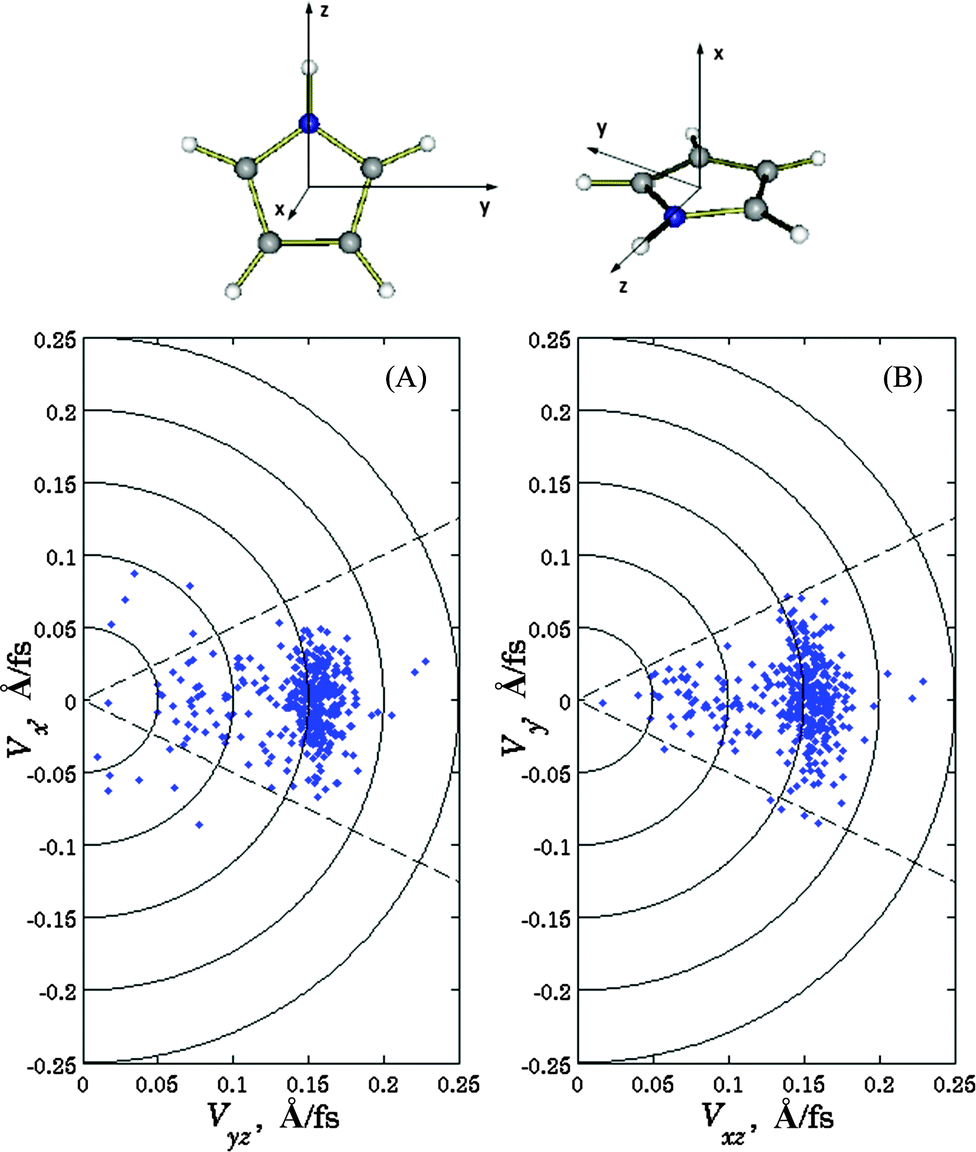 | ||
| Fig. 4 Calculated velocities of the ejected hydrogen atom with respect to the orientation of the molecule: velocities in x-direction vs. their projections on the yz plane (A), and in the y-direction vs. xz-projections (B). Definitions of the axes with respect to the molecule are shown. | ||
In order to calculate the velocity map image with respect to the laser pulse polarization, we must average the results from Fig. 3 over all possible orientations of the molecule:
I(r, φ) ∝ δ(r − |v|) ∫∫∫ dα dβ dγ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cos2(ξ(α, β, γ)) δ(φ − ϕ(θ, α, β, γ)), cos2(ξ(α, β, γ)) δ(φ − ϕ(θ, α, β, γ)), | (21) |
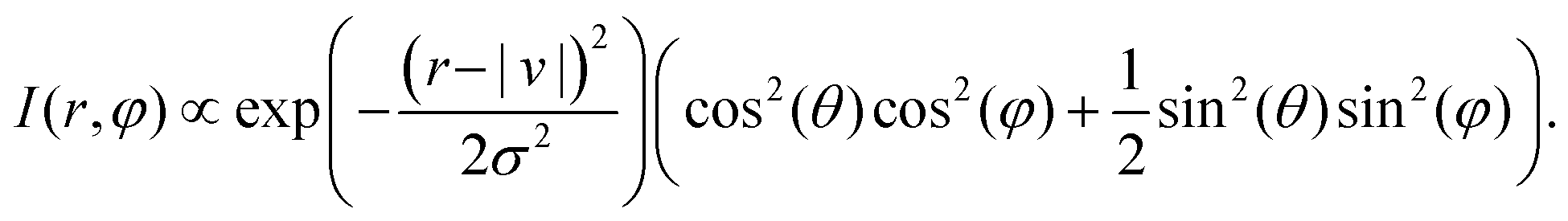 | (22) |
Fig. 5A and B show the simulated velocity map with respect to the laser pulse polarization assuming that the transition dipole is orthogonal to the N–H bond and directed along x or y directions respectively. Both frames reproduce well the main feature of the velocity map image, which is the anisotropy of the intense high energy part. Frame 5A is also consistent with experiment1a in the low energy region showing an isotropic distribution. Again, this may be an indication that the dipole moment of the transition is predominantly in the x-direction, although admittedly the statistics of both experiment and simulation are poorer in the region of low energy.
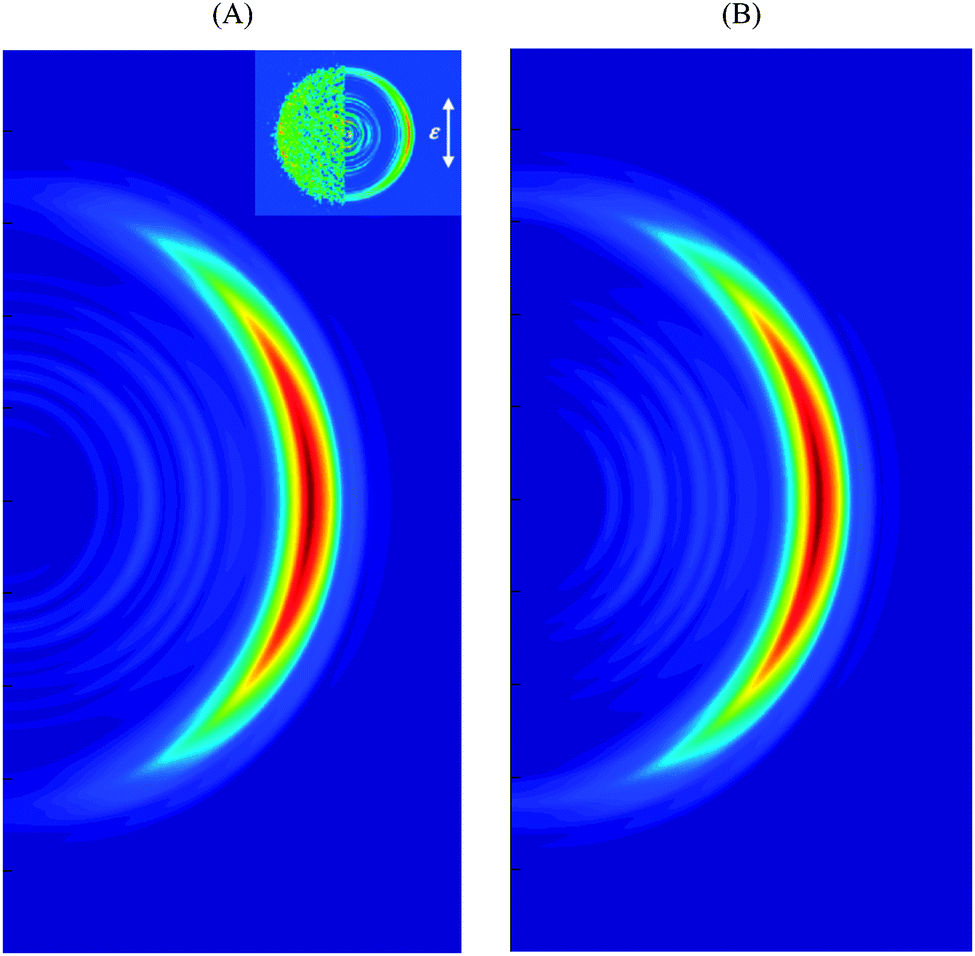 | ||
| Fig. 5 Simulated velocity map image with respect to the laser pulse polarization assuming transition dipole moment pointed in x (A) and y (B) directions. Definitions of x and y axes are given in Fig. 3. The experimental VMI1a is shown in the inset. | ||
In order to understand the character of this distribution, we plotted several typical trajectories in Fig. 6. Parts (A)–(C) show potential energy surfaces along trajectory branches, the populations, and N–H distances respectively. Fig. 6-I is an example of the trajectory with one cloning event. The dissociation is starting at about 25 fs time, when the trajectory reaches an intersection for the first time. After passing the intersection, the ground and first excited states are approximately equally populated, so the cloning procedure is applied. As one can see from Fig. 6-I (A) the potential energy for the ground state branch remains more or less constant for the rest of the run, while the excited state trajectory practically immediately passes over a potential barrier, which slows down the motion. Thus, the kinetic energies of ejected hydrogen atoms significantly differ between two branches: the ground state branch contributes to the high energy peak of the distribution in Fig. 2, while the excited state branch contributes to the low energy peak. For ground state branch, the remaining vibrational energy of the radical is low, so it remains on the ground state and does not reach the intersection again within the simulation window. The energy taken away by the hydrogen atom is lower for the excited state branch, leaving the pyrrolyl radical with sufficient energy to pass through numerous intersections and with population in both ground and excited states. The importance of intersections in the radical reached during and after dissociation has been noticed previously.8 Eventually this will result in quenching to the ground electronic state. However, this does not affect the TKER spectrum, which only monitors the dynamics until the H atom is lost.
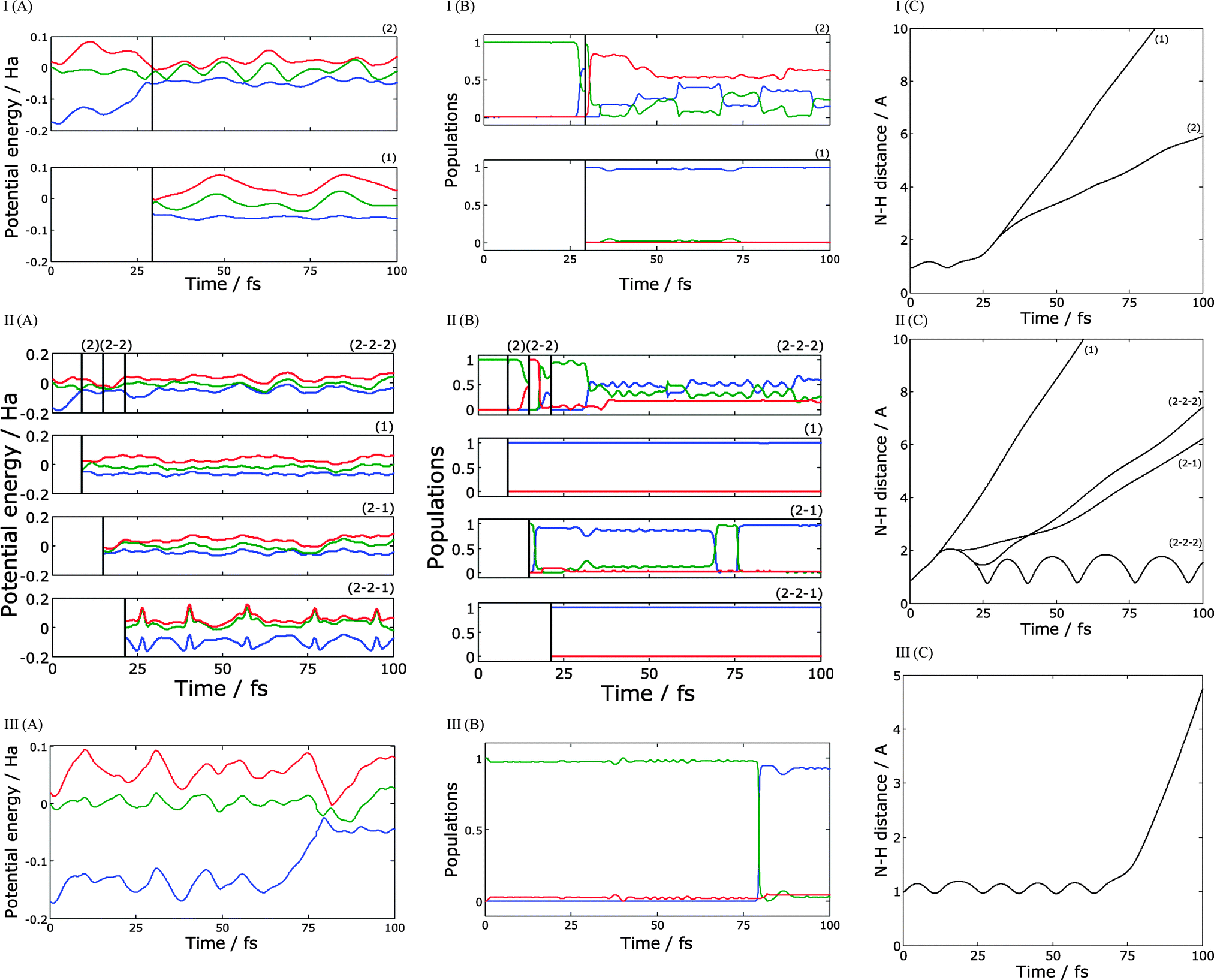 | ||
| Fig. 6 Examples of calculated trajectories with one (I), three (II), and no cloning (III): (A) – potential energy surfaces along the trajectory, (B) – the populations of electronic states, (C) – N–H distance. Populations and energies are shown for S0 (blue), S1 (green) and S2 (red) electronic states. | ||
Fig. 6-II shows a more complicated case of a trajectory with three cloning events. In this example, the trajectory reaches an intersection extremely quickly, at about 5 fs. After the cloning, ground state branch (branch 1) ejects a high-energy hydrogen atom. As in the previous example, this branch remains in the ground state and never passes through an intersection again. The excited state branch (branch 2) goes to the top of the potential barrier, where it reaches an intersection with second excited state. After another cloning, we get two new branches: branch 2–1 which is in a first excited state, and branch 2–2 in a second excited state. Branch 2–1 quickly leads to dissociation, ejecting a low-energy hydrogen atom. Branch 2–2 passes through two more intersections; the first intersection causes a practically complete transition back to the first excited state, and the second one initiates a third cloning event creating branch 2–2–1 in the ground state, and branch 2–2–2 mostly in the first excited state. Branch 2–2–2 leads to dissociation with a low energy hydrogen atom; one can see from the Fig. 6-II (C) that hydrogen speed is a little bit higher for branch 2–2–2 but still belongs to the low energy peak of the distribution in Fig. 3. For both these branches, the radical keeps sufficient energy after the dissociation and undergoes numerous transitions between the ground, first excited, and, for branch 2–2–2, second excited states. Again, these transitions are not visible in the TKER spectrum, which only probes the dynamics until H atom dissociation. Branch 2–2–1 remains in the ground state and does not lead to immediate dissociation; instead we observe high-amplitudes oscillations of N–H distance.
Fig. 6-III illustrates the simplest possible case – a trajectory without cloning. In this particular example, the N-H bond length oscillates for about 80 fs until trajectory reaches an intersection. As pointed out in Fig. 2, this is unusually long – most of the initial conditions observed to quench to the ground state (within the 200 fs simulation time) do so within the first 20–50 fs. In this case, the hydrogen atom is ejected from the ground state with high kinetic energy.
Inspection of the probabilities of electronic states did confirm the conclusion of our previous paper. Even though the radical can be formed in a mixture of electronic states with noticeable contribution of excited states, it relaxes to the ground electronic state via the intersection reached by its vibrational motion. However transitions that occur after the departure of the H atom do not affect the TKER spectrum and velocity map image calculated in this paper. Only a small fraction of dissociated trajectories retain significant contribution of excited states and forms the low kinetic energy part of the TKER spectrum and velocity map image.
V. Summary
In summary we simulated the photodissociation dynamics of pyrrole excited to the lowest singlet excited state (1A1 → 1A2) using a new efficient implementation of AIMC within the AIMS-MOLPRO code. In our implementation, the computational cost is almost the same as classical “on the fly” molecular dynamics even though the resulting dynamics is substantially quantum mechanical. This efficiency allows the accumulation of sufficient statistics to reproduce the experimental TKER spectrum and the anisotropy of the velocity map images, shedding new light on the details of the process and reaffirming our current understanding of the pyrrole photo-dynamics. Currently a number of simulations are under way in our group of the molecules such as pyrazole, imidazole previously studied experimentally.6,20Acknowledgements
DM, KS and DS acknowledge the support from EPSRC through grants EP/I014500/1 and EP/J001481/1. TJM acknowledges support from the NSF through CHE-11-24515.References
- (a) G. M. Roberts, C. A. Williams, H. Yu, A. S. Chatterley, J. D. Young, S. Ullrich and V. G. Stavros, Probing ultrafast dynamics in photoexcited pyrrole: timescales for (1)pi sigma* mediated H-atom elimination, Faraday Discuss., 2013, 163, 95–116 RSC; (b) J. Wei, J. Riedel, A. Kuczmann, F. Renth and F. Temps, Photodissociation dynamics of pyrrole: Evidence for mode specific dynamics from conical intersections, Faraday Discuss., 2004, 127, 267–282 RSC; (c) C. T. Middleton, K. de La Harpe, C. Su, Y. K. Law, C. E. Crespo-Hernandez and B. Kohler, DNA Excited-State Dynamics: From Single Bases to the Double Helix, Annu. Rev. Phys. Chem., 2009, 60(1), 217–239 CrossRef CAS PubMed.
- (a) A. L. Sobolewski and W. Domcke, Conical intersections induced by repulsive (1)pi sigma* states in planar organic molecules: malonaldehyde, pyrrole and chlorobenzene as photochemical model systems, Chem. Phys., 2000, 259(2–3), 181–191 CrossRef CAS; (b) A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Excited-state hydrogen detachment and hydrogen transfer driven by repulsive (1)pi sigma* states: A new paradigm for nonradiative decay in aromatic biomolecules, Phys. Chem. Chem. Phys., 2002, 4(7), 1093–1100 RSC; (c) Z. Lan, A. Dupays, V. Vallet, S. Mahapatra and W. Domcke, Photoinduced multi-mode quantum dynamics of pyrrole at the (1)pi sigma*-S-0 conical intersections, J. Photochem. Photobiol., A, 2007, 190(2–3), 177–189 CrossRef CAS PubMed; (d) V. Vallet, Z. G. Lan, S. Mahapatra, A. L. Sobolewski and W. Domcke, Photochemistry of pyrrole: Time-dependent quantum wave-packet description of the dynamics at the (1)pi sigma(*)-S-0 conical intersections, J. Chem. Phys., 2005, 123(14), 144307 CrossRef PubMed; (e) M. Barbatti, J. Pittner, M. Pederzoli, U. Werner, R. Mitric, V. Bonacic-Koutecky and H. Lischka, Non-adiabatic dynamics of pyrrole: Dependence of deactivation mechanisms on the excitation energy, Chem. Phys., 2010, 375(1–2), 26–34 CrossRef CAS PubMed; (f) V. Vallet, Z. Lan, S. Mahapatra, A. L. Sobolewski and W. Domcke, Time-dependent quantum wave-packet description of the 1[small pi][sigma]* photochemistry of pyrrole, Faraday Discuss., 2004, 127, 283–293 RSC; (g) Z. Lan, W. Domcke, V. Vallet, A. L. Sobolewski and S. Mahapatra, Time-dependent quantum wave-packet description of the [sup 1] pi sigma[sup *] photochemistry of phenol, J. Chem. Phys., 2005, 122(22), 224315 CrossRef PubMed; (h) S. Mahapatra, Excited Electronic States and Nonadiabatic Effects in Contemporary Chemical Dynamics, Acc. Chem. Res., 2009, 42(8), 1004–1015 CrossRef CAS PubMed.
- (a) M. N. R. Ashfold, B. Cronin, A. L. Devine, R. N. Dixon and M. G. D. Nix, The role of pi sigma* excited states in the photodissociation of heteroaromatic molecules, Science, 2006, 312(5780), 1637–1640 CrossRef CAS PubMed; (b) M. N. R. Ashfold, A. L. Devine, R. N. Dixon, G. A. King, M. G. D. Nix and T. A. A. Oliver, Exploring nuclear motion through conical intersections in the UV photodissociation of phenols and thiophenol, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(35), 12701–12706 CrossRef CAS PubMed; (c) G. A. King, A. L. Devine, M. G. D. Nix, D. E. Kelly and M. N. R. Ashfold, Near-UV photolysis of substituted phenols, Phys. Chem. Chem. Phys., 2008, 10(42), 6417–6429 RSC; (d) A. L. Devine, B. Cronin, M. G. D. Nix and M. N. R. Ashfold, High resolution photofragment translational spectroscopy studies of the near ultraviolet photolysis of imidazole, J. Chem. Phys., 2006, 125(18), 184302 CrossRef PubMed; (e) G. A. King, T. A. A. Oliver, M. G. D. Nix and M. N. R. Ashfold, Exploring the mechanisms of H atom loss in simple azoles: Ultraviolet photolysis of pyrazole and triazole, J. Chem. Phys., 2010, 132(6), 064305 CrossRef PubMed; (f) M. G. D. Nix, A. L. Devine, B. Cronin and M. N. R. Ashfold, Ultraviolet photolysis of adenine: Dissociation via the (1)pi sigma(*) state, J. Chem. Phys., 2007, 126(12), 124312 CrossRef PubMed; (g) M. G. D. Nix, A. L. Devine, B. Cronin, R. N. Dixon and M. N. R. Ashfold, High resolution photofragment translational spectroscopy studies of the near ultraviolet photolysis of phenol, J. Chem. Phys., 2006, 125(13), 133318 CrossRef PubMed; (h) A. G. Sage, M. G. D. Nix and M. N. R. Ashfold, UV photodissociation of N-methylpyrrole: The role of (1)pi sigma* states in non-hydride heteroaromatic systems, Chem. Phys., 2008, 347(1–3), 300–308 CrossRef CAS PubMed; (i) A. J. van den Brom, M. Kapelios, T. N. Kitsopoulos, N. H. Nahler, B. Cronin and M. N. R. Ashfold, Photodissociation and photoionization of pyrrole following the multiphoton excitation at 243 and 364.7 nm, Phys. Chem. Chem. Phys., 2005, 7(5), 892–899 RSC.
- (a) B. Cronin, M. G. D. Nix, R. H. Qadiri and M. N. R. Ashfold, High resolution photofragment translational spectroscopy studies of the near ultraviolet photolysis of pyrrole, Phys. Chem. Chem. Phys., 2004, 6(21), 5031–5041 RSC; (b) B. Cronin, A. L. Devine, M. G. D. Nix and M. N. R. Ashfold, Near ultraviolet photolysis of deuterated pyrrole, Phys. Chem. Chem. Phys., 2006, 8(29), 3440–3445 RSC.
- D. A. Blank, S. W. North and Y. T. Lee, The Ultraviolet Photodissociation Dynamics of Pyrrole, Chem. Phys., 1994, 187(1–2), 35–47 CrossRef CAS.
- H. Yu, N. L. Evans, V. G. Stavros and S. Ullrich, Investigation of multiple electronic excited state relaxation pathways following 200 nm photolysis of gas-phase imidazole, Phys. Chem. Chem. Phys., 2012, 14(18), 6266–6272 RSC.
- R. Montero, A. P. Conde, V. Ovejas, M. Fernandez-Fernandez, F. Castano, J. R. V. d. Aldana and A. Longarte, Femtosecond evolution of the pyrrole molecule excited in the near part of its UV spectrum, J. Chem. Phys., 2012, 137(6), 064317 CrossRef PubMed.
- K. Saita, M. G. D. Nix and D. V. Shalashilin, Simulation of ultrafast photodynamics of pyrrole with a multiconfigurational Ehrenfest method, Phys. Chem. Chem. Phys., 2013, 15(38), 16227–16235 RSC.
- (a) K. Saita and D. V. Shalashilin, On-the-fly ab initio molecular dynamics with multiconfigurational Ehrenfest method, J. Chem. Phys., 2012, 137(22), 22A506 CrossRef PubMed; (b) D. V. Shalashilin, Multiconfigurational Ehrenfest approach to quantum coherent dynamics in large molecular systems, Faraday Discuss., 2011, 153, 105–116 RSC; (c) D. V. Shalashilin, Nonadiabatic dynamics with the help of multiconfigurational Ehrenfest method: Improved theory and fully quantum 24D simulation of pyrazine, J. Chem. Phys., 2010, 132(24), 244111 CrossRef PubMed; (d) D. V. Shalashilin, Quantum mechanics with the basis set guided by Ehrenfest trajectories: Theory and application to spin-boson model, J. Chem. Phys., 2009, 130(24), 244101 CrossRef PubMed.
- P. V. Parandekar and J. C. Tully, Detailed Balance in Ehrenfest Mixed Quantum-Classical Dynamics, J. Chem. Theory Comput., 2006, 2, 229–235 CrossRef CAS.
- J. C. Tully, Molecular dyanmics with electronic transitions, J. Chem. Phys., 1990, 93, 1061–1071 CrossRef CAS PubMed.
- D. V. Makhov, W. J. Glover, T. J. Martinez and D. V. Shalashilin, Ab initio multiple cloning algorithm for quantum nonadiabatic molecular dynamics, J. Chem. Phys., 2014, 141, 054110 CrossRef PubMed.
- (a) M. Ben-Nun and T. J. Martinez, Ab Initio Quantum Molecular Dynamics, Adv. Chem. Phys., 2002, 121, 439–512 CrossRef CAS; (b) B. G. Levine and T. J. Martinez, Ab Initio Multiple Spawning Dynamics of Excited Butadiene: Role of Charge Transfer, J. Phys. Chem. A, 2009, 113(46), 12815–12824 CrossRef CAS PubMed; (c) T. J. Martinez, M. Ben-Nun and R. D. Levine, Multi-Electronic-State Molecular Dynamics: A Wave Function Approach with Applications, J. Phys. Chem., 1996, 100(19), 7884–7895 CrossRef CAS; (d) B. G. Levine, J. D. Coe, A. M. Virshup and T. J. Martinez, Implementation of ab initio multiple spawning in the Molpro quantum chemistry package, Chem. Phys., 2008, 347(1), 3–16 CrossRef CAS PubMed; (e) M. Ben-Nun and T. J. Martinez, Photodynamics of ethylene: ab initio studies of conical intersections, Chem. Phys., 2000, 259(23), 237–248 CrossRef CAS.
- D. V. Shalashilin and M. S. Child, Basis set samplng in the method of coupled coherent states: Coherent state swarms, trains and pancakes, J. Chem. Phys., 2008, 128, 054102 CrossRef PubMed.
- M. Ben-Nun and T. J. Martinez, Exploting Temporal Non-Locality to Remove Scaling Bottlenecks in Nonadiabatic Quantum Dynamics, J. Chem. Phys., 1999, 110, 4134–4140 CrossRef CAS PubMed.
- (a) D. Mendive-Tapia, B. Lasorne, G. A. Worth, M. J. Bearpark and M. A. Robb, Controlling the mechanism of fulvene S1/S0 decay: switching off the stepwise population transfer, Phys. Chem. Chem. Phys., 2010, 12(48), 15725–15733 RSC; (b) G. A. Worth, M. A. Robb and B. Lasorne, Solving the time-dependent Schrodinger equation for nuclear motion in one step: direct dynamics of non-adiabatic systems, Mol. Phys., 2008, 106(1618), 2077–2091 CrossRef CAS.
- A. J. Gianola, T. Ichino, R. L. Hoenigman, S. Kato, V. M. Bierbaum and W. C. Lineberger, Thermochemistry and electronic structure of the pyrrolyl radical, J. Phys. Chem. A, 2004, 108(46), 10326–10335 CrossRef CAS.
- A. L. Thompson, C. Punwong and T. J. Martinez, Optimization of width parameters for quantum dynamics with frozen Gaussian basis sets, Chem. Phys., 2010, 370, 70–77 CrossRef CAS PubMed.
- M. N. R. Ashfold, G. A. King, D. Murdock, M. G. D. Nix, T. A. A. Oliver and A. G. Sage, pi sigma* excited states in molecular photochemistry, Phys. Chem. Chem. Phys., 2010, 12(6), 1218–1238 RSC.
- (a) C. A. Williams, G. M. Roberts, H. Yu, N. L. Evans, S. Ullrich and V. G. Stavros, Exploring ultrafast H-atom elimination versus photofragmentation pathways in pyrazole following 200 nm excitation, J. Phys. Chem. A, 2012, 116, 2600–2609 CrossRef CAS PubMed; (b) D. J. Hadden, K. L. Wells, G. M. Roberts, L. T. Bergendahl, M. J. Paterson and V. G. Stavros, Time-resolved velocity map imaging of H-atom elimination from photoexcited imidazole and its methyl substituted derivatives, Phys. Chem. Chem. Phys., 2011, 13, 10342–10349 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp04571h |
| ‡ Current address: School of Chemistry, University of Edinburgh, West Mains Road, EH9 3JJ Edinburgh, Scotland, United Kingdom. |
| This journal is © the Owner Societies 2015 |