
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing hydrogen-bonding in binary liquid mixtures with terahertz time-domain spectroscopy: a comparison of Debye and absorption analysis†
Nicholas Y.
Tan‡
,
Ruoyu
Li‡
,
Pierre
Bräuer
,
Carmine
D'Agostino
,
Lynn F.
Gladden
and
J. Axel
Zeitler
 *
*
Department of Chemical Engineering and Biotechnology, University of Cambridge, Pembroke Street, Cambridge CB2 3RA, UK. E-mail: jaz22@cam.ac.uk; Fax: +44 (0)1223 334796; Tel: +44 (0)1223 334783
First published on 26th January 2015
Abstract
Terahertz time-domain spectroscopy is used to explore hydrogen bonding structure and dynamics in binary liquid mixtures, spanning a range of protic–protic, protic–aprotic and aprotic–aprotic systems. A direct absorption coefficient analysis is compared against more complex Debye analysis and we observed good agreement of the two methods in determining the hydrogen bonding properties when at least one of the mixture components is protic. When both components are aprotic, we show that the trend in absorption coefficients match well with the theoretical trend in strength of hydrogen bond interactions predicted based on steric and electronic properties of the components.
1. Introduction
Solutions of binary mixtures play key roles in various biological, chemical and engineering systems, such as protein folding,1,2 membrane assembly,3 electrochemical cells4 and heterogeneous catalysis.5 These binary mixtures can be broadly classified as mixtures between (1) two protic liquids, (2) an aprotic and protic liquid, and (3) two aprotic liquids. However, while the structure and dynamics of binary mixtures have been widely studied, the interaction between the liquids and its effect on the overall structure of the mixtures is not yet fully understood.Due to the importance of water in biological systems,6 protic–protic liquid mixtures that have been studied typically include water as one of the components, and systems such as water–alcohol and water–diol mixtures have been studied extensively in the past with techniques including neutron diffraction,7,8 NMR,9–13 Raman spectroscopy,14 infrared spectroscopy,15,16 dielectric spectroscopy,17 mass spectrometry18 as well as numerical and computational studies.19,20
In these systems, a larger than expected decrease in entropy and enthalpy upon mixing is observed and in early work this was attributed to an enhancement in the structuring of water molecules in the hydration shell of the solute molecule, where the water structure becomes more ice-like.21 However, recent studies have proposed alternative explanations for these observations. In particular, femtosecond infrared (fs-IR) spectroscopy measurements confirm that the “iceberg” model cannot fully explain the additional experimental data that now exists.16 Diol molecules contain two alcohol moieties, which allows the formation of intra-molecular hydrogen bonds, resulting in different conformations of diol molecules.22,23 Techniques such as conventional absorption spectroscopy24 and laser photoacoustic spectroscopy25 have been used to probe the presence of intra-molecular hydrogen bonding in diols by acquiring vapour phase OH-stretching overtone spectra of 1,3-propanediol, 1,4-butanediol and ethylene glycol. Upon mixing with water, a number of anomalous properties of diol–water mixtures have been observed with changes in mixture composition. Examples include the partial molar volume,26 the enthalpy function,27 the speed of sound in the mixtures28 and the self-diffusion coefficients of diol molecules.26 It has also been observed that isomers behave differently in aqueous mixtures,26,28,29 indicating the importance of intra-molecular hydrogen bond formation in determining the overall structure of the mixtures. Apart from aqueous mixtures, diol–alcohol mixtures have also been studied extensively, and they exhibit mixing behaviour which is closer to ideal mixing compared to the aqueous mixtures.30,31
In contrast, in the case of protic–aprotic mixtures, the aprotic molecules contain no hydroxyl or other hydrogen bond donating groups and therefore can only serve as hydrogen bond acceptors, if the appropriate functional groups are present. In particular, aqueous mixtures of aprotic systems have been the subject of extensive studies for many years and anomalous behaviours of these mixtures have been reported, such as the excess viscosity,32–35 excess molar volume,32,34,36 excess enthalpy32,37,38 and the diffusion coefficients.39 Micro-heterogeneity has been observed in these mixtures, where molecules are more likely to bond with like molecules.34,39–42 Compared to the alcohol molecules, the aprotic solvent molecules have been shown to form a smaller number of hydrogen bonds with water molecules.43–45
Finally, considering the case of aprotic–aprotic mixtures, the system of acetone and chloroform has been particularly well studied since it was found to have a negative deviation to Raoult's law,46 attributed to the preferential formation of complexes between unlike molecules.47,48 The formation of an equimolar complex between acetone and chloroform through a hydrogen bond has been confirmed both by NMR and IR studies,49–51 where chloroform molecules added to acetone were found to preferentially associate with the acetone molecules and disrupt existing acetone–acetone interactions. Other properties of this system, such as surface tension,52 activity coefficient,53 viscosity54,55 and thermodynamic excess functions,56 have also supported the formation of an acetone–chloroform complex. The translational dynamics of this system has been studied with PFG-NMR and the self-diffusion coefficients of acetone and chloroform were found to be equal in the 40% chloroform mixture,57 implying the maximal formation of acetone–chloroform complexes which move as one unit.
Despite the variety of studies performed on binary mixtures, a clear consensus on the origin of the anomalous physical properties of such solutions has not yet been reached at present, with dynamical and structural measurements often yielding apparently contradictory results. Therefore, in order to gain more insight into the structuring of binary mixtures, and transitions in their structure as a function of composition, it is necessary to employ experimental techniques that probe dynamical processes within extended hydrogen-bonded networks.
Terahertz time-domain spectroscopy (THz-TDS) is a spectroscopic technique that is able to probe the rotational and vibrational dynamics of molecules in the frequency range between 100 GHz and 4 THz.58,59 It covers part of the far-infrared region of the electromagnetic spectrum and probes molecular motions arising from inter-molecular interactions, in particular hydrogen bonds.60–63 For crystalline materials, well-defined absorption peaks are observed in the terahertz region due to the long-range order present in the crystal lattices.64–66 In contrast, the terahertz absorption spectra of liquids generally do not exhibit any distinctive absorption bands.67–69 However, due to the ability to measure both amplitude and phase of the transmitted waveform in THz-TDS, complex dielectric spectra of liquids in the terahertz range can be calculated directly from the experimental data.70–72 Dielectric relaxation analysis can then be applied to yield information on the structure and rotational dynamics of the liquids.73
The use of THz-TDS to study the structure and dynamics of hydrogen bonding in liquids has grown significantly in recent years. The technique has been used to study dielectric relaxation processes in liquid water,45,74–77 as well as the structures of acetonitrile–water44 and various alcohol–water mixtures.45,73 THz-TDS has also been extended to the study for more complex hydrogen bonded systems such as aqueous protein solutions, and has uncovered new information about the hydration shells around dissolved proteins.78
In previous studies, the applicability of THz-TDS to the study of the structure of alcohol–water mixtures has been explored.73 In particular, the relative absorption coefficients of the mixtures were shown to be indicators for non-ideal mixing and the disruption of hydrogen bond networks in the pure liquid by guest molecules. Additionally, terahertz dielectric relaxation analysis was tested, developed and discussed extensively, and a complete picture was derived of the mixing of alcohol and water molecules over the entire composition range.
In this study, we make use of the methodology developed to perform a systematic study on the structures of diol–alcohol, diol–water, aprotic solvent–water and acetone–chloroform mixtures. We demonstrate that the use of terahertz spectroscopy can be extended to study a wide range of binary liquid mixtures and show a correlation between the information obtained from the analysis of absorption coefficients and the information obtained from more complex dielectric analysis.
2. Experimental methods
2.1 Liquid samples
All liquids except for the deionised water (50 mΩ) were purchased from Sigma Aldrich or Fischer Scientific and used without further purification. All liquids were of reagent grade with ≥99% purity. Liquid mixtures were prepared by mixing the components in the appropriate ratios gravimetrically.2.2 Terahertz time-domain spectroscopy
The terahertz time-domain spectroscopy (THz-TDS) transmission setup used in this study has been described previously.73 The liquid samples were contained within a liquid cell (PIKE Technologies, Madison USA) contained between 3 mm thick z-cut quartz windows, which are transparent to terahertz radiation, separated by a 200 μm thick PTFE spacer. The sample liquid was injected through two holes drilled into the quartz window. For each sample, 200 time-domain waveforms were collected (3 min acquisition time), averaged and then transformed into the frequency domain by fast Fourier transformation (FFT). Due to the large optical mismatch between the quartz/air and quartz/sample interfaces, multiple non-negligible internal reflection pulses from the quartz window were detected. Therefore, a cut-off time before the first reflection pulse (∼43 ps) was selected prior to the FFT to eliminate any etaloning artefacts due to these multiple reflections. The multiple reflections between the window and sample interface were accounted for using the solution derived by Duvillaret et al.70 and as outlined previously.73All samples were measured at 293 ± 1 K. A sample cell with no spacer was used for the reference measurement. Data were acquired in the frequency range 0.2–2.5 THz, similar to that reported by Venables and Schmuttenmaer in their studies of mixtures of acetone, acetonitrile and methanol with water.44,45 The terahertz absorption spectra were calculated from the acquired data using the Beer–Lambert law. The complex dielectric functions were calculated from the complex refractive indices: ![[small epsilon, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e107.gif) (ω) = ε′(ω) + iε′′(ω) = [ñ(ω)]2.
(ω) = ε′(ω) + iε′′(ω) = [ñ(ω)]2.
3. Results and discussion
3.1 Diol mixtures
The experimental dielectric spectra of diol–methanol and diol–water mixtures obtained with THz-TDS were fitted to a constrained three-component Debye model (eqn (1)) as outlined in our previous discussions:73 | (1) |
One limitation of the Debye analysis is that it requires the knowledge of the static dielectric constants (εs) of each mixture. In the case of diol–methanol mixtures, these values are only available for ethylene glycol–methanol and 1,2-propanediol–methanol mixtures.30 Further, as previously mentioned, the direct physical interpretation of the relaxation times is not valid since the terahertz frequencies only cover a small range of the broad peaks described in the Debye model.
Nonetheless, the Debye model does fit the experimental data and can give an idea of the underlying physical origin of the spectral response observed at terahertz frequencies. While the quantities of τ and ε cannot be considered accurate due to the limited spectral range they are fitted to, they still represent relative changes in the relaxation behaviour.
To illustrate this, the terahertz dielectric spectra for ethylene glycol–methanol and 1,2-propanediol–methanol mixtures were fitted with Debye model, and the calculated relaxation strengths are shown in Fig. 1. Fig. 1 also shows the theoretical ideal relaxation strengths of bulk methanol and bulk diol based on an ideal mixing model where the liquid molecules preserve their rotational dynamics from the pure liquids. As such, their ideal dielectric relaxation strengths are the picosecond relaxation strength of the pure liquid weighted by its volume fraction in the mixture.73 This analysis holds true for the systems studied here as they exhibit negligible excess volumes of mixing.
 | ||
| Fig. 1 Relative relaxation strengths of diol–methanol mixtures obtained using the three-component Debye model (ε1, ε2, ε3) and ideal values εmethanol, εdiol (dashed lines) for (a) ethylene glycol–methanol and (b) 1,2-propanediol–methanol mixtures. Previously reported εs values were used for fitting.30 | ||
For the methanol–diol mixtures, the fitted values of ε1 and ε2 agree well with the ideal values, indicating that these mixtures exhibit relatively ideal mixing. This agrees with previous reports that mixtures of methanol and similar alcohols such as ethanol and ethylene glycol can be considered ideal, and can be explained by the alignment of hydrophilic and hydrophobic groups which minimise the disruption to the original hydrogen bonding network.80 In the case of diol–water mixtures, ε1 is found to be consistently lower than the ideal values of εwater, while ε2 is consistently higher than the ideal values of εdiol (Fig. 2), which is similar to the results found for monoalcohol–water mixtures.73
 | ||
| Fig. 2 Relative relaxation strengths of diol–water mixtures obtained using the three-component Debye model (ε1, ε2, ε3) and ideal values εwater, εdiol (dashed lines) for (a) ethylene glycol–water, (b) 1,2-propanediol–water, (c) 1,3-propanediol–water, (d) 1,2-butanediol–water, and (e) 1,4-butanediol–water mixtures. Previously reported εs values were used for fitting.28,81 | ||
The lower values of ε1 compared to εwater can be attributed to the disruption of the water structure by the diol molecules, while the higher values of ε2 compared to εdiol are most likely due to the incorporation of both bulk diol and diol–water structures into this component of the model.
It follows that ε2 − εdiol represents the extent of hydrogen bonded diol–water networks in the mixtures. When plotted against the diol mole fractions (Fig. 3), it is found that the values of ε2 − εdiol, and hence degree of hydrogen bond interactions between diol and water, have a maximum (Xmax) at 30% for ethylene glycol mixtures, 20% for both propanediol isomer mixtures, and 10% for both butanediol isomer mixtures. This suggests the presence of two distinct regimes in these mixtures: (1) a water rich regime (xdiol < Xmax) where each additional diol molecule interacts with water and disrupts the bulk water structure and (2) a diol rich regime (xdiol > Xmax) where each additional diol molecule interacts with other diol molecules and bulk diol structures begin to form.
 | ||
| Fig. 3 ε 2 − εdiol plotted over the entire concentration range of various diol–water mixtures: (a) ethylene glycol–water, (b) 1,2-propanediol–water, (c) 1,3-propanediol–water, (d) 1,2-butanediol–water, (e) 1,4-butanediol–water. | ||
Apart from the analysis carried out on the THz dielectric spectra, it is also possible to extract information about the degree of interaction between the components of a binary mixture by studying the relative changes in the absorption coefficient.
The representative terahertz absorption spectra of pure liquids, ethylene glycol–water and ethylene glycol–methanol mixtures are shown in Fig. 4. The absorption spectra are featureless and increase linearly with increasing frequency, and are similar across the range of diols studied. Given that the absorption of water and methanol are higher than that of the diols, the overall absorption of the mixture decreases with increasing diol concentration.
 | ||
| Fig. 4 THz absorption of (a) water–ethylene glycol mixtures, (b) methanol–ethylene glycol mixtures. | ||
As discussed previously,73 the non-ideal behaviour of binary mixtures can be described by
| αrelative = αideal − αreal | (2) |
| αideal(ω) = X1α1(ω) + X2α2(ω) | (3) |
Fig. 5 shows the relative absorption coefficients of five diol–methanol and five diol–water mixtures over the entire concentration range for each mixture. As the spectra increase monotonously with frequency and the spectral response does not change significantly with varying liquid composition, the absorption coefficients were averaged over 3 frequency points spanning the accessible frequency range to better reflect the broadband spectral response instead of artificially choosing a single frequency.73αrelative represents the average of the relative absorption coefficients at 0.5, 1.0 and 1.5 THz.
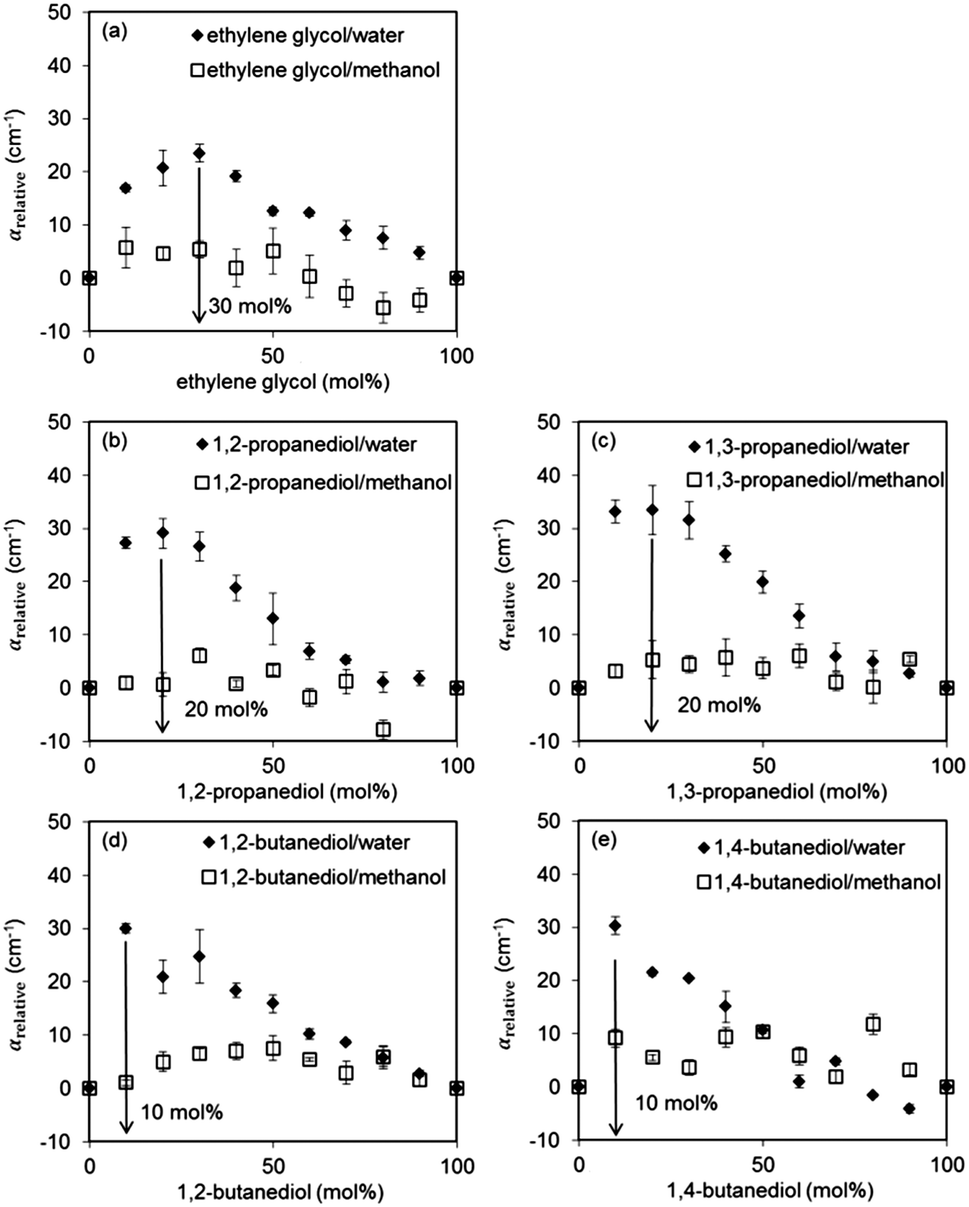 | ||
| Fig. 5 Relative THz absorption coefficients for different alcohol–water–methanol mixtures: (a) ethylene glycol–water–methanol, (b) 1,2-propanediol–water–methanol, (c) 1,3-propanediol–water–methanol, (d) 1,2-butanediol–water–methanol, (e) 1,4-butanediol–water–methanol. | ||
It is apparent from Fig. 5 that the effect of non-ideal mixing in these mixtures results in a decrease in the terahertz absorption (i.e. αrelative takes positive values, see eqn (2)). These results are consistent with previously reported data for other binary mixtures44 and can be attributed to the disruption of existing hydrogen bond networks in the pure liquids by the introduction of guest molecules, where the disruption of existing hydrogen bonds results in the formation of new hydrogen bond networks between the pure liquid and the guest molecules that have slower rotational dynamics than the original pure liquids, therefore reducing the terahertz absorption.
The decrease in absorption could also arise from a decrease in the amount of hydrogen bonding in the mixtures. However, when the THz dielectric spectra of these mixtures are fitted with a two-component Debye model, the picosecond relaxation times, which represent the overall rotational dynamics of the mixtures, are found to increase with increasing diol concentration, indicating that the introduction of the guest diol molecules does indeed slow down the cooperative rotational dynamics of the mixtures (see ESI†).
It can also be observed that the diol–methanol mixtures behave closer to ideal mixtures than diol–water mixtures, since αrelative ≈ 0. This is in agreement with the fitted dielectric data obtained using the constrained three-component Debye model and supports the suggestion that diol–methanol mixtures can be considered ideal. In contrast, diol–water mixtures exhibit clearly non-ideal behaviour since the presence of hydrophobic aliphatic groups disrupts the water structure through hydrophobic hydration. The diol mole fractions where the mixtures are most non-ideal are 30% for ethylene glycol, 20% for both propanediol isomers and 10% for both butanediol isomers. As the size of the aliphatic group of the diol increases, so does the effect of hydrophobic hydration, which results in maximum non-ideality at a lower diol concentration. Isomerism of propanediol and butanediol does not affect the degree of non-ideality of the mixtures, which implies that intra-molecular hydrogen bonding is not a contributing factor to the terahertz absorption as the diol hydroxyl groups preferentially form inter-molecular hydrogen bonds with water. However, it is shown that the maximum value of αrelative is lower in ethylene glycol–water mixtures compared to other diol–water mixtures. This indicates a lesser degree of non-ideality and is possibly due to the preferential formation of a five-membered ring species through an intra-molecular hydrogen bond, decreasing the extent of interactions between the diol and water.
It is striking that the concentrations at which non-ideality is observed in the αrelative analysis are identical to those obtained from the earlier Debye analysis. This implies that both methods are equally effective in characterising the deviation in non-ideal behaviour of these liquid mixtures. Since, as already stated, the interpretation of the Debye analysis is limited as a result of only being able to fit to the narrow THz spectral range, we now consider only the absorption analysis for other systems, since this approach is both simple and robust.
3.2 Aprotic solvent–water mixtures
It has been shown that aprotic liquids exhibit broad liberational bands in their terahertz spectra centred in the 1.5 to 3.0 THz range.82 As a consequence, it proved to be much more difficult to establish an accurate dielectric model to fit the data for aprotic liquid–water mixtures compared to the diol–water mixtures due to the influence of these liberational bands.As described above, a two-component Debye model is first used to fit the THz dielectric spectra to obtain the values of τ for the pure components in the mixture so that the constrained three-component Debye model can be used. However, in order to account for the liberational bands of the aprotic solvents, a one-component Debye model with a vibrational term would be a more appropriate model of the system (eqn (4)). Therefore, the experimental data were fitted to both models and tested for their fits.
 | (4) |
It was found that the values associated with the picosecond dielectric relaxation process, ε1 and τ1, were similar for both models. However, eqn (4) only fitted the data well at high concentrations of aprotic liquid (≥80%) for acetone–water and tetrahydrofuran–water mixtures. For the acetonitrile–water mixtures, both models gave similarly good fits since the liberational band of acetonitrile lies beyond the dynamic range of this experiment45 (ωv = 2.79 THz, see ESI†).
In aprotic liquids, the picosecond dielectric relaxation process occurs faster than in water (Fig. 6), τ1(aprotic) < τ1(water), because of the significantly weaker dipole–dipole interactions between the aprotic molecules. For the acetone–water and tetrahydrofuran–water mixtures, τ1 increases rapidly as aprotic liquid is added to water before gradually decreasing. This suggests that adding acetone or tetrahydrofuran to water results in an enhancement of structure due to the formation of hydrogen bonds between the two components of the mixtures, which is characterised by slower rotational dynamics. Since τ1 reaches a maximum at 20% acetone and 10% tetrahydrofuran, we can conclude that the interactions between the aprotic solvents and water are the greatest at these concentrations.
 | ||
| Fig. 6 Relative relaxation times of the picosecond relaxation component (τ1) for (a) acetonitrile–water, (b) acetone–water, (c) tetrahydrofuran–water mixtures. D2 model refers to the two-component Debye model, while the D1-V1 model refers to the model described in eqn (4). | ||
For acetonitrile–water mixtures, τ1 decreases monotonously with increasing acetonitrile concentration. This implies that acetonitrile is less efficient at disrupting the structure of water. It has been found that acetonitrile molecules form less hydrogen bonds with water molecules and there is a high number of acetonitrile molecules in these mixtures which do not accept hydrogen bonds.45
When the constrained three-component Debye model was applied to these systems, it was found to perform well across all concentrations for the acetonitrile–water system, but only above 50% aprotic solvent for the acetone–water and tetrahydrofuran–water system (see ESI†). Due to the failure of the constrained three-component Debye model in fully describing the acetone and tetrahydrofuran–water systems, we proceeded with further analysis on only the acetonitrile–water system.
Due to the faster picosecond relaxation of acetonitrile compared to water, the dielectric relaxation process due to acetonitrile–water interactions is now convoluted with the relaxation of the bulk liquid with slower rotational dynamics, water (ε1, Fig. 7). Therefore, it is the value ε1 − εwater that is representative of the hydrogen bonded acetonitrile–water structures, in contrast to ε2 − εdiol in diol–water mixtures. As seen in Fig. 7, ε1 − εwater has a broad and shallow maximum from 20–60% acetonitrile, in contrast to the sharp maxima found in other systems. This is consistent with previous reports of micro-heterogeneity in this concentration range for acetonitrile–water mixtures.32–34,39,43,85 As such, the water molecules preferentially self-associate, so the relative degree of hydrogen bonding between acetonitrile and water remains relatively constant despite an increasing number of acetonitrile molecules.
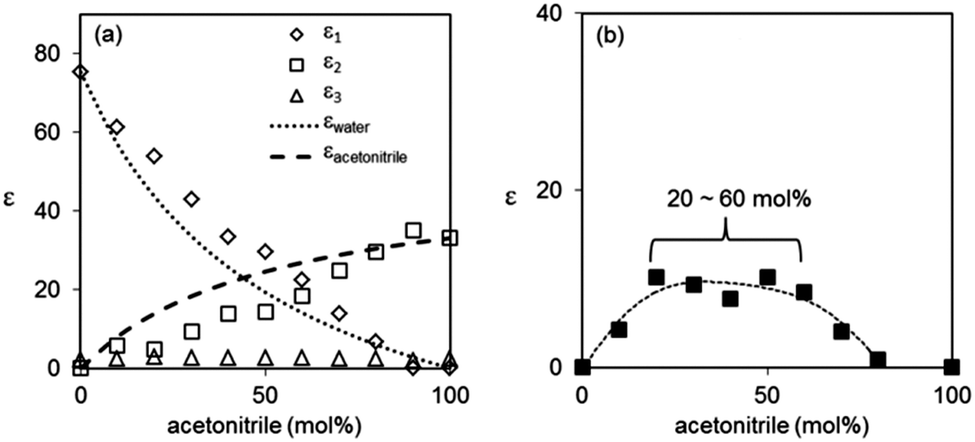 | ||
| Fig. 7 (a) Relative relaxation strengths of acetonitrile–water mixtures evaluated with the three-component Debye model (ε1, ε2, ε3) and ideal values for εwater and εacetonitrile (dashed lines). (b) ε1 − εwater for varying concentrations of acetonitrile–water mixtures. Dashed line in (b) plotted to guide the eye. Previously reported εs value was used for fitting.83 | ||
The representative THz absorption spectra for the aprotic liquids–water mixtures are shown in Fig. 8. Due to the presence of liberational bands in the aprotic solvents as mentioned previously, the absorption spectra of aprotic liquids–water mixtures also exhibit these liberational bands as the concentration of the aprotic liquid increases. The liberational band for acetonitrile is centred at about 3.0 THz, which is outside the dynamic range of the current experiment, therefore it is not observed in acetonitrile–water mixtures.
 | ||
| Fig. 8 Representative terahertz absorption spectra of aprotic liquid–water mixtures: (a) acetonitrile–water, (b) acetone–water and (c) tetrahydrofuran–water. | ||
As before, the relative absorption coefficients for each mixture were calculated at 0.5, 1.0 and 1.5 THz and these were averaged to obtain αrelative. The maximum values of αrelative correspond to the maximum non-ideality in these mixtures and occur at the following concentrations (Xmax): 20–50% acetonitrile, 20% acetone and 10% tetrahydrofuran in their respective mixtures with water (Fig. 9). Due to the strong absorption of acetonitrile–water mixtures in the terahertz region, the experimental uncertainty for this system was relatively large, precluding the accurate determination of a single maximum point.
 | ||
| Fig. 9 Relative THz absorption coefficients for different aprotic liquid–water mixtures: (a) acetonitrile–water, (b) acetone–water, (c) tetrahydrofuran–water. Dashed line in (a) plotted to guide the eye. | ||
At these concentrations, previous studies have reported anomalies in various properties of these mixtures, including viscosity,32–35 excess molar volume,32,34,36 excess enthalpy32,37,38 and diffusion coefficients,39 which are likely related to hydrogen bond formation between water and aprotic liquid molecules and support the terahertz data.
Since the trends in αrelative are a consequence of hydrogen bonding and there is only one possible hydrogen bond acceptor–donor pair in these systems, the variation in Xmax across the three mixtures can be explained through the relative basicities of the aprotic liquids. The relative Lewis basicities can be expressed via the BF3 affinities of these molecules, which is the enthalpy change of the complexation reaction with BF3. Based on BF3 affinities, tetrahydrofuran is most basic, followed by acetone, with acetonitrile the least basic.86 This suggests that tetrahydrofuran has the greatest tendency to form hydrogen bonds with water, hence rapidly interacts with water and disrupts the bulk water structure, resulting in maximum non-ideality at the lowest concentration of aprotic liquid. For the acetonitrile–water system, not only does it require a greater concentration of aprotic liquid to reach maximum non-ideality, the absolute maximum of αrelative is the lowest of the three systems, indicating the lowest degree of interaction with water. As with the diol–methanol and diol–water systems, the values of Xmax obtained with the absorption analysis of these systems matches closely with the concentrations at which dielectric analysis shows greatest cross species interaction in the mixtures. While the Debye analysis has the potential to provide useful quantitative information regarding these systems, its utility is limited when applied only to THz data. When only THz data is studied, the spectral range is too small for the Debye analysis to provide quantitative information of the relaxation strengths and times. As such, the Debye analysis only provides information regarding the relative differences between systems, which can also be achieved with the analysis of αrelative. In addition, the Debye analysis is constrained by two other factors: (1) the need to find an appropriate model to describe each specific system, which becomes more complex as aprotic components are introduced, and (2) the need to have known values for εs. Both of these issues have been highlighted above, and the former becomes a major hurdle when aprotic solvents are studied.
The analysis of the absorption of binary mixtures through αrelative has consistently demonstrated its ability to investigate non-ideal behaviour and provide information on the relative affinity of a solute for the pure solvent, as well as the extent of hydrogen bonding between the two components of the mixture. Given that the Debye analysis does not add any significant information to the αrelative analysis when studying isolated terahertz data, it is not used for subsequent systems, which were selected to further investigate the general utility of the αrelative analysis.
3.3 Aprotic liquid–aprotic liquid mixtures
In mixtures of acetone–chloroform, two liberational bands are observed, arising from each of the aprotic liquids. At high chloroform concentrations, the liberational band is observed around 1 THz, corresponding to that of chloroform. However, the spectra rapidly shift towards a liberational band at 1.8 THz, corresponding to acetone, due to the significantly more strongly absorbing acetone dominating the spectra (Fig. 10).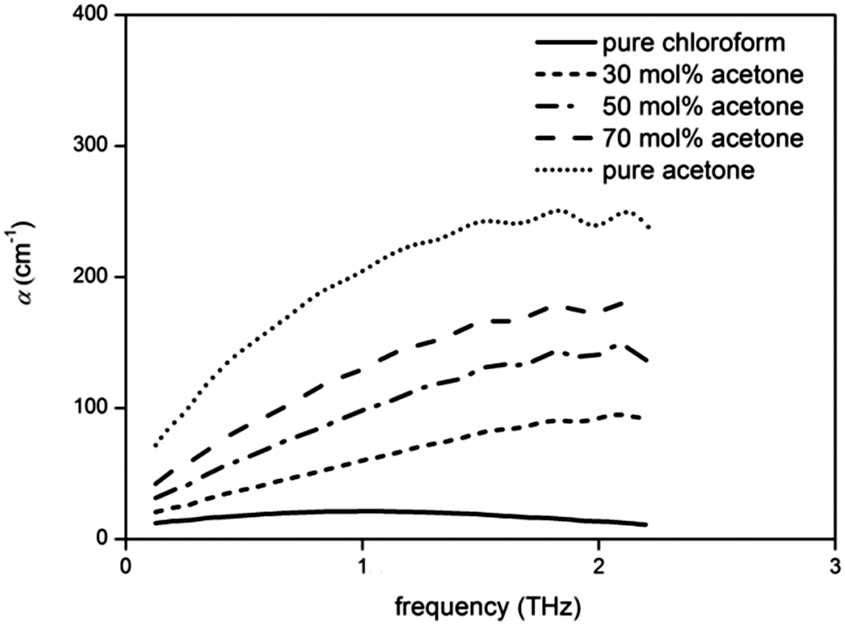 | ||
| Fig. 10 Representative terahertz absorption spectra of acetone–chloroform mixtures. | ||
Analysis of the relative absorption coefficient of acetone–chloroform mixtures shows that they exhibit similar trends compared to diol–water and aprotic liquid–water mixtures, where αrelative increases as acetone concentration increases, reaching a maximum at Xmax = 43% acetone (Fig. 11). The maximum value of αrelative is also found to be lower than for diol–water and aprotic liquid–water mixtures, which implies that the degree of interaction between acetone–chloroform is lower than that of the other systems, either due to weaker hydrogen bonds or less hydrogen bonds. As previously discussed, acetone and chloroform have the tendency to form complexes via a hydrogen bond interaction between the chloroform hydrogen and carbonyl oxygen in acetone.47–51 However, given that the hydrogen bond donor in this system is not directly bonded to an electronegative oxygen, it is relatively weaker compared to diol–water and aprotic–water mixtures. For comparison, the acetone–chloroform hydrogen bond strength87 is estimated to be 8.8 kJ mol−1, while the hydrogen bond strength in water88 is approximately 23.3 kJ mol−1. Additionally, the extent of hydrogen bonding in this system will be smaller due to lower number of hydrogen bond donors and acceptors.
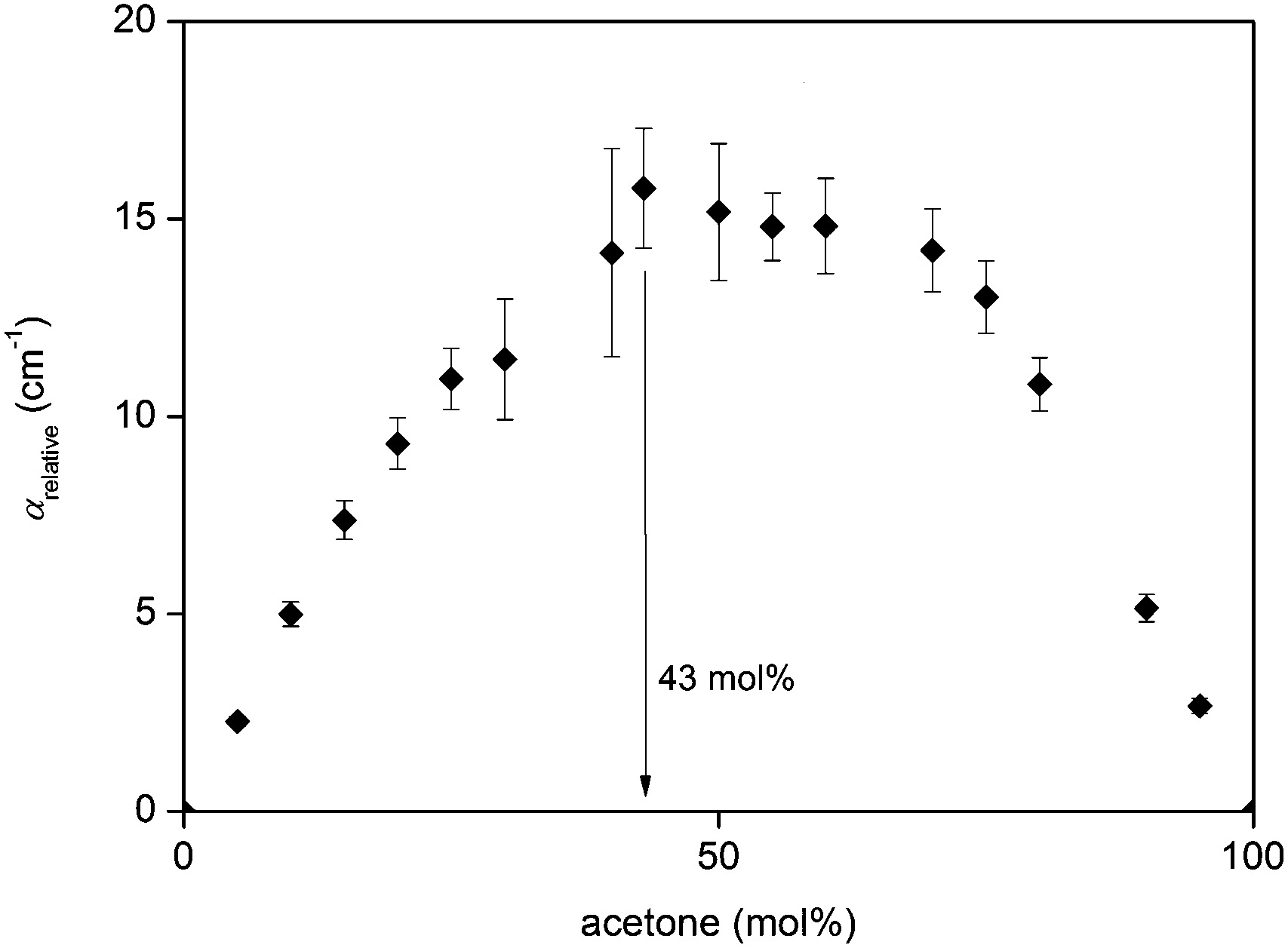 | ||
| Fig. 11 Relative THz absorption coefficients for acetone–chloroform mixtures across the entire range of concentrations. | ||
1H NMR and thermodynamic studies have shown that maximum complex formation occurs around 40% acetone.56,57 In 1H NMR experiments, the self-diffusion coefficients of acetone and chloroform are equal at 40% acetone, and it is proposed that this is when hydrogen bonding between the two species is at its greatest, hence they diffuse as one complex.57 This corresponds to the Xmax seen in the THz absorption data, where there is greatest interaction between acetone and chloroform, resulting in the slowest rotational dynamics in the system. However, more recent investigations show conflicting data and do not report the presence of such a point where the self-diffusion coefficients are equal.89 While it may be tempting to directly compare the NMR diffusion data and the THz absorption analysis, one must be cautious to note that the NMR data primarily describe translational dynamics, but the THz data primarily describe rotational dynamics, so direct comparisons of the data obtained from both techniques may not be accurate.
In order to further probe the utility of THz absorption analysis, mixtures of acetone derivatives with chloroform were studied with THz-TDS. The selected acetone derivatives were 3-pentanone, 2,4-dimethyl-3-pentanone and chloroacetone. The introduction of additional methyl groups in 3-pentanone and 2,4-dimethyl-3-pentanone increases the steric hindrance around the hydrogen bond acceptor. In addition, the basicities of these derivatives decrease as the number of methyl groups is increased.86,90 Given that αrelative represents the decrease in absorption due to interactions between the two components of binary mixtures, the maximum value of αrelative should be indicative of the extent of hydrogen bonding between the components. In addition, the concentration at which αrelative reaches a maximum should be directly related to the ease of hydrogen bond formation between the two components of the mixture.
Since the strength of hydrogen bond interactions is dependent on the basicity of the hydrogen bond acceptor, the predicted trend for the maxima in αrelative is acetone > 3-pentanone > 2,4-dimethyl-3-pentanone. However, the carbonyl oxygen is less accessible with additional methyl groups and therefore maximum complex formation should occur at higher concentrations of the acetone derivatives, with the predicted trend for Xmax being 2,4-dimethyl-3-pentanone > 3-pentanone > acetone.
In chloroacetone, one of the hydrogen atoms is substituted for chlorine. This increases the steric hindrance around the carbonyl oxygen and also exerts a significant electronic effect. It has been shown via vibrational spectroscopy91 and BF3 affinity measurements86 that substitution of hydrogen with chlorine reduces the basicity of the carbonyl group due to the electron-withdrawing effects of chlorine. With reduced basicity of the hydrogen bond acceptor, chloroacetone should interact less strongly with chloroform. Therefore, chloroacetone–chloroform mixtures should behave much more ideally than acetone–chloroform mixtures, with the absolute maximum of αrelative being much lower. Due to the increased steric hindrance from the chlorine atom, the Xmax for chloroacetone–chloroform mixtures should also be at higher chloroacetone concentrations compared to acetone–chloroform.
Fig. 12 shows the αrelative plots for all three acetone derivatives and chloroform mixtures. It can be seen that the experimental trends match well with the predicted trends based on the varying steric and electronic effects of the acetone derivatives. This shows that terahertz absorption analysis is a valid tool for studying the relative affinity of the components of hydrogen bonding binary mixtures, which is observed through the relative position of Xmax, as well as the extent of hydrogen bonding, which is observed through the relative values of αrelative at Xmax.
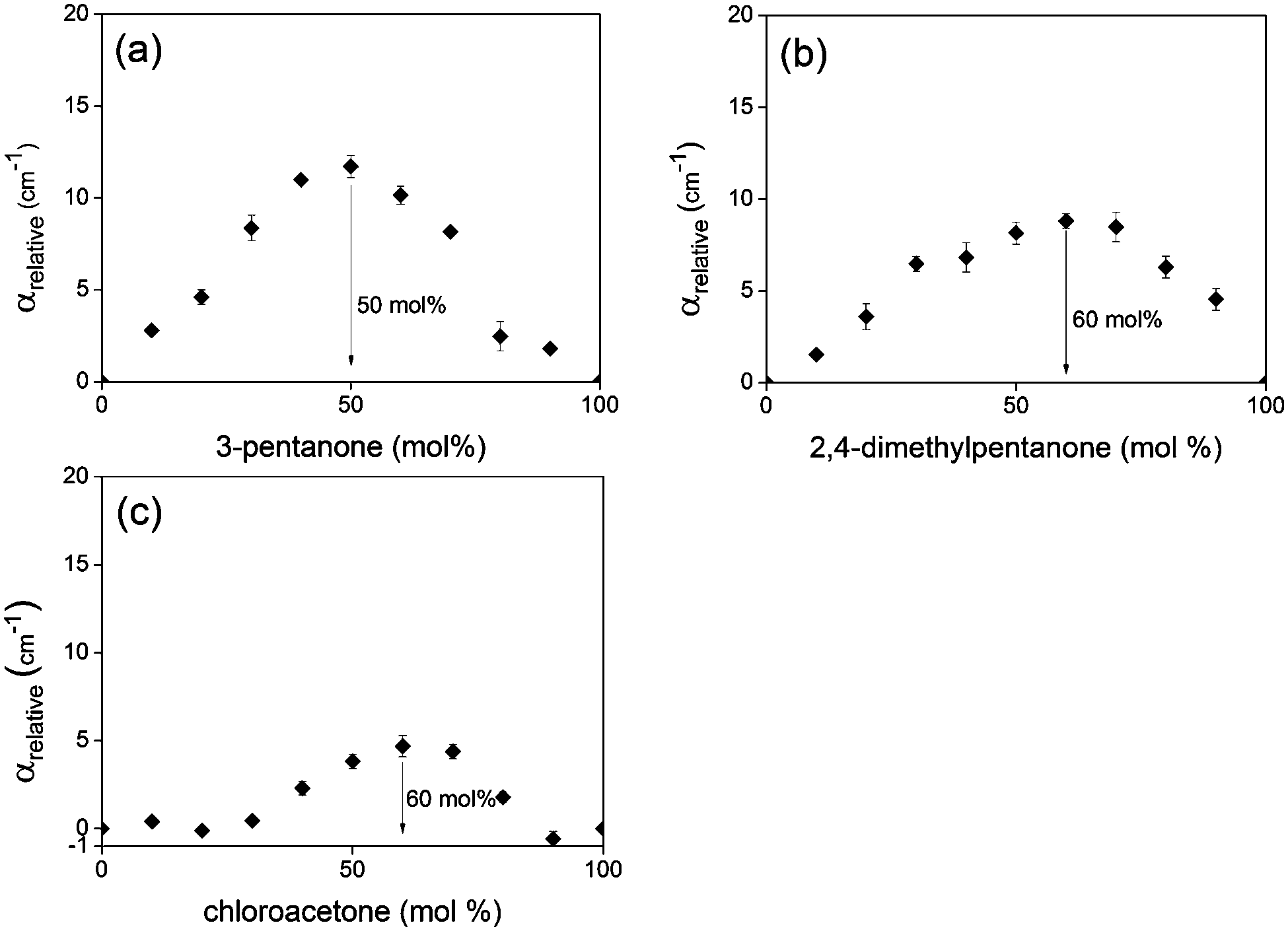 | ||
| Fig. 12 Relative THz absorption coefficients across the entire range of concentrations for (a) 3-pentanone–chloroform, (b) 2,4-dimethyl-3-pentanone–chloroform, (c) chloroacetone–chloroform mixtures. | ||
4. Conclusion
Terahertz spectroscopy was used to study a range of binary mixtures, which fall within three broad categories (diol–water, aprotic liquid–water and aprotic liquid–aprotic liquid) to investigate hydrogen bonding in these systems. Terahertz measurements allowed for the extraction of the absorption coefficients of the various binary mixtures at different concentrations. In addition, the dielectric relaxation strengths and time constants could be extracted from the terahertz data. Relative absorption coefficient analysis of these systems was found to be both simple and robust to perform, but still provided valuable information about the non-ideality in these systems arising from hydrogen bonding, allowing for the concentration at which there is maximum hydrogen bonding interaction between liquids to be determined. In addition, the absolute value of the relative absorption coefficient provides a measure of the relative strength of hydrogen bonding interactions between the two components in the binary mixture and allows for the comparison of hydrogen bonding between different binary mixture systems.While Debye analysis has been used to analyse such systems in the past and as part of this study, we found that it is limited by the currently accessible spectral range probed by terahertz spectroscopy as well as the need to find appropriate model equations that accurately represent the relaxational/vibrational characteristics for each system. Development of accurate models to describe the relaxation processes in binary mixtures not trivial, particularly when aprotic liquids make up one or both mixture components due to additional complications arising from the liberational bands of these liquids. Therefore, the relative absorption coefficient analysis is a valuable alternative to Debye analysis as it (1) is reliable across a spectrum of binary mixtures, (2) does not rely on trial and error to find the most suitable model, (3) does not require any additional information. With these three criteria in mind, this method would be well-suited to analysing the interaction of more complex mixtures even in the presence of heterogeneous catalysts and can be extended to the real-time analysis of solvent systems in large reactors for chemical engineering.
Acknowledgements
The authors would like to acknowledge funding provided by EPSRC Grant EP/G011397/1.Notes and references
- E. V. Kudryashova, A. K. Gladilin, A. V. Vakurov, F. Heitz, A. V. Levashov and V. V. Mozhaev, Biotechnol. Bioeng., 1997, 55, 267–277 CrossRef CAS.
- A. Cammers-Goodwin, T. J. Allen, S. L. Oslick, K. F. McClure, J. H. Lee and D. S. Kemp, J. Am. Chem. Soc., 1996, 118, 3082–3090 CrossRef CAS.
- N. Kimizuka and T. Nakashima, Langmuir, 2001, 17, 6759–6761 CrossRef CAS.
- J.-P. Belieres, D. Gervasio and C. A. Angell, Chem. Commun., 2006, 4799–4801 RSC.
- B. S. Akpa, C. D'Agostino, L. F. Gladden, K. Hindle, H. Manyar, J. McGregor, R. Li, M. Neurock, N. Sinha, E. H. Stitt, D. Weber, J. A. Zeitler and D. W. Rooney, J. Catal., 2012, 289, 30–41 CrossRef CAS PubMed.
- P. Ball, Nature, 2008, 452, 291–292 CrossRef CAS PubMed.
- L. Dougan, R. Hargreaves, S. P. Bates, J. L. Finney, V. Réat, A. K. Soper and J. Crain, J. Chem. Phys., 2005, 122, 174514 CrossRef CAS PubMed.
- S. Dixit, J. Crain, W. C. K. Poon, J. L. Finney and A. K. Soper, Nature, 2002, 416, 829–832 CrossRef CAS PubMed.
- C. Corsaro, J. Spooren, C. Branca, N. Leone, M. Broccio, C. Kim, S.-H. Chen, H. E. Stanley and F. Mallamace, J. Phys. Chem. B, 2008, 112, 10449–10454 CrossRef CAS PubMed.
- Y. Ishihara, S. Okouchi and H. Uedaira, J. Chem. Soc., Faraday Trans., 1997, 93, 3337–3342 RSC.
- A. Coccia, P. L. Indovina, F. Podo and V. Viti, J. Chem. Phys., 1975, 7, 30–40 CAS.
- C. D'Agostino, G. L. Brett, P. J. Miedziak, D. W. Knight, G. J. Hutchings, L. F. Gladden and M. D. Mantle, Chem. – Eur. J., 2012, 18, 14426–14433 CrossRef PubMed.
- C. D'Agostino, T. Kotionova, J. Mitchell, P. J. Miedziak, D. W. Knight, S. H. Taylor, G. J. Hutchings, L. F. Gladden and M. D. Mantle, Chem. – Eur. J., 2013, 19, 11725–11732 CrossRef PubMed.
- M. Nedic, T. N. Wassermann, R. Wugt Larsen and M. A. Suhm, Phys. Chem. Chem. Phys., 2011, 13, 14050–14063 RSC.
- N. Nishi, S. Takahashi, M. Matsumoto, A. Tanaka, K. Muraya, T. Takamuku and T. Yamaguchi, J. Phys. Chem., 1995, 99, 462–468 CrossRef CAS.
- Y. L. A. Rezus and H. J. Bakker, Phys. Rev. Lett., 2007, 99, 148301 CrossRef CAS.
- T. Sato, A. Chiba and R. Nozaki, J. Chem. Phys., 1999, 110, 2508–2521 CrossRef CAS PubMed.
- A. Wakisaka, K. Matsuura, M. Uranaga, T. Sekimoto and M. Takahashi, J. Mol. Liq., 2011, 160, 103–108 CrossRef CAS PubMed.
- S. M. Mejía, E. Flórez and F. Mondragón, J. Chem. Phys., 2012, 136, 144306 CrossRef PubMed.
- F. Biscay, A. Ghoufi and P. Malfreyt, J. Chem. Phys., 2011, 134, 044709 CrossRef CAS PubMed.
- H. S. Frank and M. W. Evans, J. Chem. Phys., 1945, 13, 507–532 CrossRef CAS PubMed.
- M. Mandado, R. A. Mosquera and C. Van Alsenoy, Tetrahedron, 2006, 62, 4243–4252 CrossRef CAS PubMed.
- W. Caminati, J. Mol. Spectrosc., 1981, 86, 193–201 CrossRef CAS.
- D. L. Howard, P. Jorgensen and H. G. Kjaergaard, J. Am. Chem. Soc., 2005, 127, 17096–17103 CrossRef CAS PubMed.
- D. L. Howard and H. G. Kjaergaard, J. Phys. Chem. A, 2006, 110, 10245–10250 CrossRef CAS PubMed.
- H. Piekarski, M. Jozwiak, J. Woznicka, A. Bald and A. Szejgis, Phys. Chem. Liq., 1995, 30, 195–207 CrossRef CAS.
- A. M. Zaichikov, J. Struct. Chem., 2007, 48, 94–103 CrossRef CAS.
- J. George and N. V. Sastry, J. Chem. Eng. Data, 2003, 48, 1529–1539 CrossRef CAS.
- V. Zhuravlev and T. Usacheva, Moscow Univ. Chem. Bull., 2010, 65, 225–228 CrossRef.
- R. J. Sengwa, S. Sankhla and N. Shinyashiki, J. Solution Chem., 2008, 37, 137–153 CrossRef CAS.
- F. F. Hanna, B. Gestblom and A. Soliman, J. Mol. Liq., 2002, 95, 27–40 CrossRef CAS.
- M. I. Davis, Thermochim. Acta, 1983, 63, 67–82 CrossRef CAS.
- M. I. Davis, Thermochim. Acta, 1984, 73, 149–164 CrossRef CAS.
- C. Moreau and G. Douheret, Thermochim. Acta, 1975, 13, 385–392 CrossRef CAS.
- A. C. Kumbharkhane, S. N. Helambe, M. P. Lokhande, S. Doraiswamy and S. C. Mehrotra, Pramana, 1996, 46, 91–98 CrossRef CAS.
- Y. P. Handa and G. C. Benson, J. Solution Chem., 1981, 10, 291–300 CrossRef CAS.
- O. Kiyohara, P. J. Darcy and G. C. Benson, Can. J. Chem., 1979, 57, 1006–1010 CrossRef CAS PubMed.
- D. N. Glew and H. Watts, Can. J. Chem., 1973, 51, 1933–1940 CrossRef CAS.
- E. V. Goldamme and H. G. Hertz, J. Phys. Chem., 1970, 74, 3734 CrossRef.
- Y. Marcus and Y. Migron, J. Phys. Chem., 1991, 95, 400–406 CrossRef CAS.
- Y. Migron and Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 1339–1343 RSC.
- H. Kovacs and A. Laaksonen, J. Am. Chem. Soc., 1991, 113, 5596–5605 CrossRef CAS.
- J. E. Bertie and Z. D. Lan, J. Phys. Chem. B, 1997, 101, 4111–4119 CrossRef CAS.
- D. S. Venables and C. A. Schmuttenmaer, J. Chem. Phys., 1998, 108, 4935–4944 CrossRef CAS PubMed.
- D. S. Venables and C. A. Schmuttenmaer, J. Chem. Phys., 2000, 113, 11222–11236 CrossRef CAS PubMed.
- J. Zawidzki, Z. Phys. Chem., 1900, 35, 129–203 Search PubMed.
- F. Dolezalek, Z. Phys. Chem., 1908, 64, 727–748 Search PubMed.
- A. Apelblat, A. Tamir and M. Wagner, Fluid Phase Equilib., 1980, 4, 229–255 CrossRef CAS.
- C. M. Huggins, G. C. Pimentel and J. N. Shoolery, J. Chem. Phys., 1955, 23, 1244–1247 CrossRef CAS PubMed.
- K. B. Whetsel and R. E. Kagarise, Spectrochim. Acta, 1962, 18, 329–339 CrossRef CAS.
- V. A. Durov and I. Y. Shilov, J. Chem. Soc., Faraday Trans., 1996, 92, 3559–3563 RSC.
- H. P. Meissner and A. S. Michaels, Ind. Eng. Chem., 1949, 41, 2782–2787 CrossRef CAS.
- A. E. Karr, E. G. Scheibel, W. M. Rowes and D. F. Othmer, Ind. Eng. Chem., 1951, 43, 961–968 CrossRef CAS.
- R. J. Fort and W. R. Moore, Trans. Faraday Soc., 1966, 62, 1112–1119 RSC.
- A. E. Karr, W. M. Rowes and E. G. Scheibel, Anal. Chem., 1951, 23, 459–463 CrossRef CAS.
- E. R. Kearns, J. Phys. Chem., 1961, 65, 314–316 CrossRef CAS.
- D. W. McCall and D. C. Douglass, J. Phys. Chem., 1967, 71, 987–997 CrossRef CAS.
- M. Vanexter, C. Fattinger and D. Grischkowsky, Opt. Lett., 1989, 14, 1128–1130 CrossRef CAS.
- C. A. Schmuttenmaer, Chem. Rev., 2004, 104, 1759–1779 CrossRef CAS PubMed.
- M. Walther, B. Fischer, M. Schall, H. Helm and P. U. Jepsen, Chem. Phys. Lett., 2000, 332, 389–395 CrossRef CAS.
- J. A. Zeitler, P. F. Taday, M. Pepper and T. Rades, J. Pharm. Sci., 2007, 96, 2703–2709 CrossRef CAS PubMed.
- J. A. Zeitler, P. F. Taday, K. C. Gordon, M. Pepper and T. Rades, ChemPhysChem, 2007, 8, 1924–1927 CrossRef CAS PubMed.
- M. Tonouchi, Nat. Photonics, 2007, 1, 97–105 CrossRef CAS.
- R. Y. Li, J. A. Zeitler, D. Tomerini, E. P. J. Parrott, L. F. Gladden and G. M. Day, Phys. Chem. Chem. Phys., 2010, 12, 5329–5340 RSC.
- M. Takahashi, Y. Kawazoe, Y. Ishikawa and H. Ito, Chem. Phys. Lett., 2009, 479, 211–217 CrossRef CAS PubMed.
- D. G. Allis, D. A. Prokhorova and T. M. Korter, J. Phys. Chem. A, 2006, 110, 1951–1959 CrossRef CAS PubMed.
- D. Grischkowsky, S. Keiding, M. Vanexter and C. Fattinger, J. Opt. Soc. Am. B, 1990, 7, 2006–2015 CrossRef CAS.
- M. Naftaly and R. E. Miles, J. Appl. Phys., 2007, 102, 6 CrossRef PubMed.
- S. N. Taraskin, S. I. Simdyankin, S. R. Elliott, J. R. Neilson and T. Lo, Phys. Rev. Lett., 2006, 97, 055504 CrossRef CAS.
- L. Duvillaret, F. Garet and J. L. Coutaz, Appl. Opt., 1999, 38, 409–415 CrossRef CAS.
- I. Pupeza, R. Wilk and M. Koch, Opt. Express, 2007, 15, 4335–4350 CrossRef CAS.
- E. P. J. Parrott, J. A. Zeitler, L. F. Gladden, S. N. Taraskin and S. R. Elliott, J. Non-Cryst. Solids, 2009, 355, 1824–1827 CrossRef CAS PubMed.
- R. Li, C. D'Agostino, J. McGregor, M. D. Mantle, J. A. Zeitler and L. F. Gladden, J. Phys. Chem. B, 2014, 118, 10156–10166 CrossRef CAS PubMed.
- C. Rønne, L. Thrane, P. O. Astrand, A. Wallqvist, K. V. Mikkelsen and S. R. Keiding, J. Chem. Phys., 1997, 107, 5319–5331 CrossRef PubMed.
- D. A. Schmidt, O. Birer, S. Funkner, B. P. Born, R. Gnanasekaran, G. W. Schwaab, D. M. Leitner and M. Havenith, J. Am. Chem. Soc., 2009, 131, 18512–18517 CrossRef CAS PubMed.
- K. J. Tielrooij, D. Paparo, L. Piatkowski, H. J. Bakker and M. Bonn, Biophys. J., 2009, 97, 2484–2492 CrossRef CAS PubMed.
- H. Yada, M. Nagai and K. Tanaka, Chem. Phys. Lett., 2008, 464, 166–170 CrossRef CAS PubMed.
- S. Ebbinghaus, S. J. Kim, M. Heyden, X. Yu, U. Heugen, M. Gruebele, D. M. Leitner and M. Havenith, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20749–20752 CrossRef CAS PubMed.
- R. Barthel, K. Bachhuber, R. Buchner and H. Hetzenauer, Chem. Phys. Lett., 1990, 165, 369–373 CrossRef.
- J. Ortega, C. Rafols, E. Bosch and M. Roses, J. Chem. Soc., Perkin Trans. 2, 1996, 1497–1503 RSC.
- J. George and N. V. Sastry, Fluid Phase Equilib., 2004, 216, 307–321 CrossRef CAS PubMed.
- R. Li, Hydrogen Bonding Structure and Dynamics Studied by Terahertz Time-Domain Spectroscopy, PhD thesis, University of Cambridge, UK, 2012 Search PubMed.
- C. Moreau and G. Douheret, J. Chem. Thermodyn., 1976, 8, 403–410 CrossRef CAS.
- F. E. Critchfield, J. A. Gibson and J. L. Hall, J. Am. Chem. Soc., 1953, 75, 6044–6045 CrossRef CAS.
- A. J. Easteal, Aust. J. Chem., 1980, 33, 1667–1675 CrossRef CAS.
- C. Laurence and J.-F. Gal, Lewis Basicity and Affinity Scales: Data and Measurement, John Wiley & Sons, Ltd, Chichester, UK, 2009 Search PubMed.
- A. N. Campbell and E. M. Kartzmark, Can. J. Chem., 1960, 38, 652–655 CrossRef CAS.
- S. J. Suresh and V. M. Naik, J. Chem. Phys., 2000, 113, 9727–9732 CrossRef CAS PubMed.
- C. D'Agostino, J. A. Stephens, J. D. Parkinson, M. D. Mantle, L. F. Gladden and G. D. Moggridge, Chem. Eng. Sci., 2013, 95, 43–47 CrossRef PubMed.
- P. C. Maria, J. F. Gal, J. D. Franceschi and E. Fargin, J. Am. Chem. Soc., 1987, 109, 483–492 CrossRef CAS.
- W. Gordy and S. C. Stanford, J. Phys. Chem., 1941, 9, 204–214 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp04477k |
| ‡ N.Y.T. and R.L. contributed equally to this work. |
| This journal is © the Owner Societies 2015 |