
Fullerene cyanation does not always increase electron affinity: an experimental and theoretical study
Tyler T.
Clikeman
a,
Shihu H. M.
Deng
b,
Alexey A.
Popov
*c,
Xue-Bin
Wang
*b,
Steven H.
Strauss
*a and
Olga V.
Boltalina
*a
aDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523, USA. E-mail: steven.strauss@colostate.edu; olga.boltalina@colostate.edu; Fax: +1-970-491-1801; Tel: +1-970-491-5088
bPhysical Sciences Division, Pacific Northwest National Laboratory, P.O. Box 999, MS K8-88, Richland, Washington 99352, USA
cDepartment of Electrochemistry and Conducting Polymers, Leibniz Institute for Solid State and Materials Research, 01069 Dresden, Germany
First published on 7th November 2014
Abstract
The electron affinities of C70 derivatives with trifluoromethyl, methyl and cyano groups were studied experimentally and theoretically using low-temperature photoelectron spectroscopy (LT PES) and density functional theory (DFT). The electronic effects of these functional groups were determined and found to be highly dependent on the addition patterns. Substitution of CF3 for CN for the same addition pattern increases the experimental electron affinity by 70 meV per substitution. The synthesis of a new fullerene derivative, C70(CF3)10(CN)2, is reported for the first time.
Introduction
Introduction of electron-withdrawing groups (EWGs) into organic molecules is known to generally enhance their electron accepting properties, and this approach has been actively used to design new n-type organic semiconductor materials.1,2 Among organic electronic devices, organic photovoltaics (OPVs) are regarded as commercially viable renewable energy sources that have already achieved efficiencies over 10% by utilizing conducting polymers and fullerenes as electron-donors and electron-acceptors, respectively.3 Energy level engineering of donor-acceptor pairs directly affects the optimal driving force for maximum charge transfer in OPV active layers.4 As new low-band-gap polymers are synthesized, new fullerene molecules with increased electron accepting strength (higher electron affinity (EA) or deeper LUMO levels) must follow in order to match the frontier orbitals of the donor and achieve the optimal driving force for maximizing charge transfer. Modification of the fullerene core with EWGs has been widely used to tune E(LUMO) levels and achieve better OPV performance. For example, C60(CF3)25 and C60(CN)26 were reported as favorable alternatives to PCBM and C60(indene) fullerene cycloadducts in certain active layer formulations. Furthermore, an electron-withdrawing CN moiety was intentionally attached to the C60(indene) acceptor to match the orbital energies of a low-band-gap polymer, and OPV device performance showed improved power conversion efficiency compared to underivatized C60(indene).7 Cyanation of trifluoromethylated C60 resulted in large increases in EA for the C60R5 structures with a skew pentagonal pyramid addition motif.8 Direct comparison of electron-withdrawing effects of CF3 and CN groups on E(LUMO) in the isostructural compounds revealed that cyanation is more potent than trifluoromethylation.9Herein we report several examples of cyanated C70 derivatives for which an opposite effect on EA upon addition of CN groups was observed. Therefore, one cannot consider cyanation as a general and straightforward approach for boosting acceptor properties in fullerenes and their derivatives.
Results and discussion
General remarks
In the research community developing OPV materials and optimizing device performance, organic molecule acceptors are commonly referred to as materials with low-lying LUMO levels (i.e., lowest unoccupied molecular orbitals). The quantitative value of LUMO energy has been conveniently (but not always consistently and correctly)10 derived from the measured reversible first reduction potential (E1/2 value) using cyclic voltammetry with typical uncertainties of 10–20 mV. Determination of solid-state EA of organic materials is now being performed via inverse photoelectron spectroscopy (IPES), but with lower precision and accuracy; errors as high as 300 meV are typically reported.11,12 Gas-phase EA measured by low-temperature photoelectron spectroscopy (LT PES) represents the most fundamental intrinsic measure of a molecule's ability to accept electrons, and uncertainties as low as 5–10 meV can be readily achieved in modern state-of-the-art instruments, allowing for reliable comparisons of compounds even with very small differences in EA, for example C60 and C70.13,14 This method is indispensable when organic acceptors do not exhibit reversible electrochemical behavior in solution, and when the amounts of materials are too small to be used for IPES which is frequently the case at the exploratory stage of research.In this work we studied the electronic properties of new C70(CF3)n(CN)m fullerene acceptors using LT PES and DFT calculations. Their synthesis and structural characterization have been published recently; however, electrochemical properties were not reported because of electrochemical irreversibility and insufficient sample amounts for some minor products.39 An original conception of the project was to develop more powerful acceptors than trifluoromethylfullerenes (TMFs) by adding more electron-withdrawing CN groups to TMF substrates. Our theoretical E(LUMO) predictions9 as well as experimental evidence reported by us8 and others7,15 for the cyanated C60 derivatives indicated that a considerable enhancement of acceptor properties occurs when CN groups are attached to a parent C60, C60(CF3)2n or C60(indene). Furthermore, gas-phase studies of C70(CN)n species by electrospray mass spectrometry accompanied by semi-empirical theoretical analysis demonstrated the propensity of C70 to form stable singly- and doubly-charged C70(CN)n anions, where n = 1–6.16 No bulk samples of C70(CN)n compounds have been reported in the literature, to our knowledge.
The family of C70(CF3)n compounds that were chosen as substrates for cyanation in this study currently includes dozens of structurally and spectroscopically characterized molecules, some of which can be prepared selectively and in large quantities.17,18 Several isomers of C70(CF3)n possess unique photophysical properties, i.e., they were proven to be the brightest fluorophores of all fullerenes.19 Electronic properties of C70 TMFs were studied by cyclic voltammetry and theoretically using the DFT method, see Fig. 1 for DFT calculated E(LUMO) for C70(CF3)n.17
 | ||
| Fig. 1 DFT calculated E(LUMO) for known C70(CF3)n (n = 0–12) compounds.17 | ||
These studies revealed that all but one C70(CF3)n molecule have significantly lower-lying LUMOs than the parent C70, with 0.218 < ΔE(LUMO, vs. C70) < 0.514, and thus are much better acceptors than parent C70. The E(LUMO) shift of only 0.036 eV was calculated for the most abundant isomer of C70(CF3)10 (denoted here as 10-1, where 10 is the number of CF3 groups, 1 is the isomer number in C70(CF3)10, similar notations are used for other isomers throughout the text).† Such an unexpected result was in line with the observed small negative shift in E1/2vs. C70. In the case of all the other C70(CF3)n compounds, large positive shifts in E1/2vs. C70 were measured, and up to three quasi-reversible reductions were recorded. It was found that 8-2 had the highest positive shift in E1/2 as well as the lowest-lying LUMO in the entire series (Fig. 1).17
For the studies of the cyanation effect on electronic properties of C70 TMFs, two C70(CF3)n substrates were chosen, Cs-C70(CF3)8 (8-1) and C1-C70(CF3)10 (10-1). Both compounds represent the most thermodynamically stable isomers among respective compositions, they form most abundantly in high-temperature syntheses, and thus can be readily prepared and isolated in practical amounts following previously reported literature procedures.17,18 Notably, 10-1 contains the 8-1 substructure plus the addition of 2 CF3 groups (see Fig. 2 for Schlegel diagrams).
 | ||
| Fig. 2 Schlegel diagrams of C70 TMFs and their cyanated derivatives studied by the DFT method. Filled and open circles represent cage carbon atoms of bonded CF3 and CN, respectively, triangles represent the location of CH3 addition. Yellow indicates a continuous ribbon of edge sharing p-, m-, and o-C6X2 hexagons. meta and ortho C6X2 hexagons are labeled with m and o, respectively. | ||
Electron affinity of C70(CF3)8 and C70(CF3)10
First, we determined gas-phase EAs of 8-1 and 10-1. Fig. 3 (bottom panel) and Fig. 4 (bottom panel) show the photoelectron spectra of the mono-anions of 8-1 and 10-1, respectively, recorded at 266 nm (4.661 eV) using acetonitrile solutions of each fullerene mixed with appropriate donors using a magnetic-bottle time-of-flight PES coupled with an ESI source and a cryogenic ion-trap for size-selected anions as described previously.20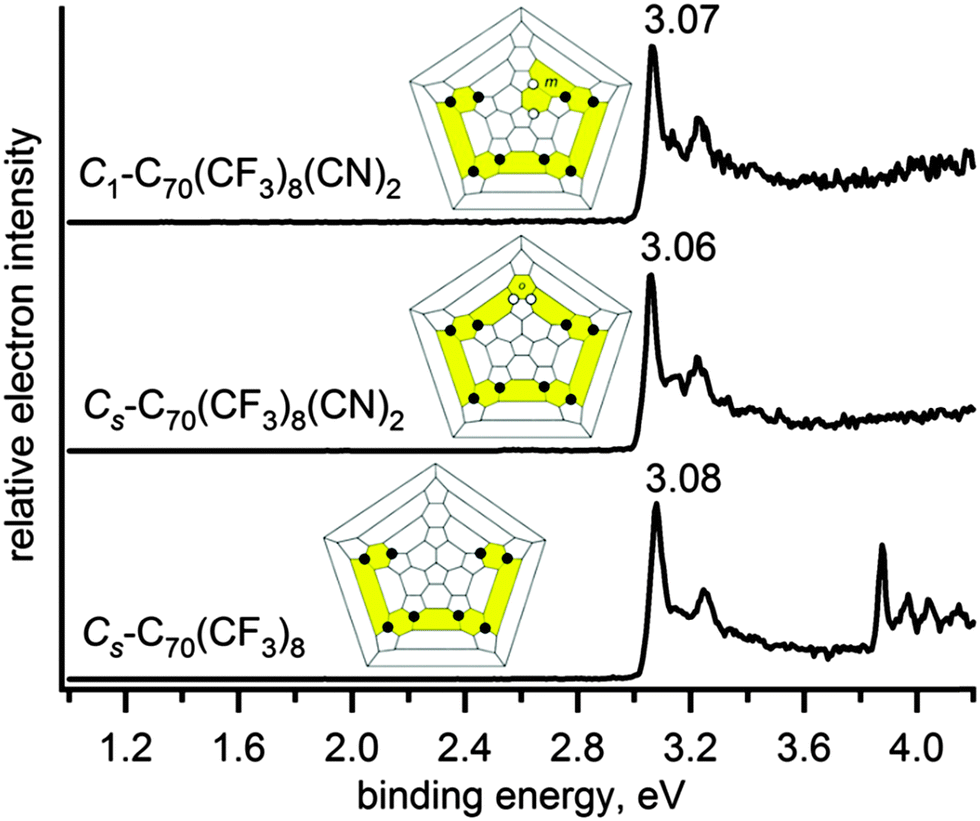 | ||
| Fig. 3 The low temperature (12 K) photoelectron spectra at 266 nm of Cs-C70(CF3)8 (8-1), Cs-C70(CF3)8(CN)2 (Cs-(8-1)(CN)2), and C1-C70(CF3)8(CN)2 (C1-(8-1)(CN)2). | ||
 | ||
| Fig. 4 The low temperature (12 K) photoelectron spectra at 266 nm of C1-C70(CF3)10 (10-1), C1-C70(CF3)10(CN)2(CH3)2-a ((10-1)(CN)2(CH3)2-a), C1-C70(CF3)10(CN)2(CH3)2-b ((10-1)(CN)2(CH3)2-b), and C1-C70(CF3)10(CN)2 ((10-1)(CN2)). | ||
The experimentally determined adiabatic EA of 8-1 is 3.08(1) eV which is 0.315 eV higher than the EA value of parent C70 measured using the PES technique,14 whereas the EA value measured here for 10-1 is only 2.93(1) eV. A 150 meV decrease in the EA of 10-1 compared to 8-1 is likely caused by the destabilization of the fullerene π system due to the introduction of two additional CF3 groups into 8-1 structure, and it is not compensated by the electron-withdrawing effect of the CF3 groups. These measured EA values are in qualitative agreement with the previously calculated E(LUMO) for these molecules, i.e., 8-1 was predicted to have a 0.204 eV lower-lying LUMO than 10-1. We also calculated their EA values by the DFT method in this work, which agree well with the experiment: the relative differences in EA between 8-1 and 10-1 are ΔEADFT = 0.202 eV. This validation of the DFT-derived EA values was further used for the comparison of the electronic properties for the pair of C2-C70(CF3)8 (8-2) and C2-C70(CF3)10 (10-2) compounds, the latter has the 8-2 substructure in its addition pattern as shown in the Schlegel diagrams, Fig. 2. The 8-2 molecule was predicted to be a stronger acceptor than 10-2 based on E(LUMO) values and reduction potentials,17 which is now supported by comparing their new DFT data for EAs: EADFT(8-2) = 3.224 eV, which is 0.272 eV higher than that of 10-2. Remarkably, 8-2 appears to be an even stronger acceptor than F4-TCNQ,21 and comparable to fluorinated fullerenes, C60F18 and C60F36,22 the latter has been widely used as a p-dopant in organic electronics,23–25 and for enhancement of diamond surface conductivity.26 This makes 8-2 a promising alternative to a fluorofullerene for doping applications, not only due to comparable EA values, but also because it is chemically more inert towards hydrolysis.27,28
Electron affinity of cyanated C70(CF3)8
We then investigated if the destabilization resulting from two CF3 additions to 8-1 as observed in 10-1 could be overcome by adding stronger electron-accepting groups than CF3, and whether the locations of these EWGs on the cage influence the EAs. Two products of cyanation of 8-1 were prepared for this work using a recently published method,39Cs-(8-1)(CN)2 and C1-(8-1)(CN)2. They were studied by LT PES, and the resulting spectra, together with the Schlegel diagrams, are shown in Fig. 3. These results reveal that introducing two CN groups into 8-1 does not actually increase the EA in these two cyanated derivatives, as one might expect, but it even results in slight decreases of 10–20 meV. The strong electron-withdrawing nature of CN groups was not enough to overcome destabilization caused by the changed π system for these two isomers. These slight decreases in EA compared to parent 8-1 were confirmed by DFT calculated EA values as shown in Table 1.| Compound | Abbreviation | Experimental EAa (eV) | DFT EA (eV) |
|---|---|---|---|
| a Uncertainty is ±10 meV. b See ref. 14. c See ref. 31. | |||
| C70 | C70 | 2.765(1)b, 2.72(5)c | 2.522 |
| C s-C70(CF3)8 | 8-1 | 3.08 | 2.934 |
| C 2-C70(CF3)8 | 8-2 | Not measured | 3.224 |
| C s -C70(CF3)8(CN)2 | C s-(8-1)(CN)2 | 3.06 | 2.892 |
| C 1-C70(CF3)8(CN)2 | C 1-(8-1)(CN)2 | 3.07 | 2.909 |
| C 1-C70(CF3)8(CN)2-th | C 1-(8-1)(CN)2-th | n/a | 3.147 |
| C 1-C70(CF3)10 | 10-1 | 2.93 | 2.732 |
| C 2-C70(CF3)10 | 10-2 | Not measured | 2.952 |
| C 1-C70(CF3)10(CN)2 | (10-1)(CN)2 | 3.14 | 3.000 |
| C 1-C70(CF3)10(CN)2(CH3)2-a | (10-1)(CN)2(CH3)2-a | 2.97 | 2.849 |
| C 1-C70(CF3)10(CN)2(CH3)2-b | (10-1)(CN)2(CH3)2-b | 2.97 | 2.850 |
To examine if placing CN group(s) on different cage carbon atoms in 8-1 may cause an opposite effect, i.e., lead to an increased EA value, we used DFT to study a theoretical isomer of C70(CF3)8(CN)2 (C1-(8-1)(CN)2-th) that has the same addition pattern as 10-2 and only differs from Cs-(8-1)(CN)2 by the location of one CN moiety (see Fig. 2). Indeed, this minimal change in the addition pattern leads to a large increase in the EA of 0.213 eV compared to 8-1 (Table 1). Thus, the introduction of two CN groups into 8-1 can either increase or decrease EA based on the addition pattern. The calculated EAs of three isomers of C70(CF3)8(CN)2 differ by as much as 0.255 eV, which manifests strong dependence of electronic properties on the location of each substituent on the fullerene core. In contrast, small-molecule acceptors with polycyclic aromatic cores exhibit linear incremental dependence on the number of EWGs, and isomeric molecules within the same composition preserve very similar EAs.29,30
Our results indicate that the relative electron-withdrawing effect of a CN group is larger than that of the CF3 group when ipso-substitution of a CF3 group takes place. For pairs of isostructural C70(CF3)8(CN)2 and C70(CF3)10 with the 10-1 and 10-2 addition patterns, incremental increase in DFT-calculated EA(per one CN group) is ca. 90 meV and 100 meV, respectively, which is in agreement with the recently reported incremental EA value of 80 meV per one CN group for the isostructural pair of C60(CF3)5 and C60(CF3)4CN species.8 Experimental data from photoelectron spectroscopy reported here provide an unequivocal support for these conclusions derived from the DFT data: the measured EA of C1-(8-1)(CN)2 is 140 meV higher than that of 10-1, indicating that substituting CF3 for CN results in an increase of EA by 70 meV per substitution. Furthermore, the C70(CF3)8(CN)2 and C70(CF3)10 isomers with the 10-2 addition pattern have an increased EA over the 10-1 pattern by 238 meV and 220 meV, respectively.
Electron affinity of cyanated C70(CF3)10
The electronic properties of the three cyano derivatives of 10-1 were also studied by LT PES and DFT. Two isomers of C1-C70(CF3)10(CN)2(CH3)2 ((10-1)(CN)2(CH3)2-a and (10-1)(CN)2(CH3)2-b) were synthesized using a previously reported procedure.39 A new compound, C1-C70(CF3)10(CN)2 ((10-1)(CN2)), was prepared from 10-1 according to a modified literature procedure (see the footnote for complete experimental details).†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39 The addition of NEt4CN to 10-1 resulted in one predominant isomer of C1-C70(CF3)10(CN)− that was subsequently quenched with p-TsCN to produce one predominant product, (10-1)(CN)2. The 19F NMR spectrum of this new fullerene exhibited ten CF3 multipets that differ from the ten multiplets of 10-1 as shown in Fig. 5, and is practically identical to the 19F NMR spectra of the previously characterized derivative C1-C70(CF3)10(CN)(CH3).39 Therefore, the new derivative most likely has the same addition pattern as the latter, where two cyano groups are added to the most reactive cage carbons, as shown in Fig. 5.
39 The addition of NEt4CN to 10-1 resulted in one predominant isomer of C1-C70(CF3)10(CN)− that was subsequently quenched with p-TsCN to produce one predominant product, (10-1)(CN)2. The 19F NMR spectrum of this new fullerene exhibited ten CF3 multipets that differ from the ten multiplets of 10-1 as shown in Fig. 5, and is practically identical to the 19F NMR spectra of the previously characterized derivative C1-C70(CF3)10(CN)(CH3).39 Therefore, the new derivative most likely has the same addition pattern as the latter, where two cyano groups are added to the most reactive cage carbons, as shown in Fig. 5.
 | ||
| Fig. 5 The fluorine-19 NMR spectra of 10-1 and (10-1)(CN)2 and corresponding Schlegel diagrams. | ||
The gas-phase EA of the newly synthesized (10-1)(CN)2 was determined by PES and compared to 10-1. Cyanation of 10-1 resulted in a 210 meV increase of EA. Remarkably, comparison of 10-1 and two isomers of C1-C70(CF3)10(CN)2(CH3)2 for which photoelectron spectroscopy data were also obtained in this work shows slightly higher EA values for the latter despite the presence of two electron-donating CH3 groups in their structures and the decreased π-system of the C70 core. The measured EA values of two C1-C70(CF3)10(CN)2(CH3)2 isomers are virtually the same: apparently, the difference in the relative positions of CN and CH3 does not have an effect on their electronic properties. DFT calculations confirm the observed trend of increasing EA from 10-1 to (10-1)(CN)2(CH3)2 to (10-1)(CN)2 (Table 1 and Fig. 4). In contrast to the unanticipated trends in the electronic properties of cyanated C70(CF3)8 compounds described above, these three examples of chemical modifications of 10-1 appear to follow the expected trend where electron-withdrawing CN groups increase EA and electron-donating CH3 groups decrease EA.
Conclusions
We have studied the effects of trifluoromethylation and cyanation on the electron affinity of seven C70 fullerene derivatives using LT PES and DFT. In some cases, the addition of electron-withdrawing CF3 or CN groups leads to the improvement in electron accepting properties, whereas cyanation led to decreased EA in several other cases. In the case of two fullerene derivatives with the same addition pattern, CN substitution of CF3 results in an EA increase of 70 meV per substitution, as determined experimentally, and an increase of 90-100 meV was predicted by DFT. DFT calculations on two different C70(CF3)8(CN)2 isomers demonstrated that the difference in the location of only one CN group can change the EA by 255 meV. Overall, excellent correlation between the experimentally determined and DFT calculated EA values (with systematic underestimation of DFT values of ca. 150 meV) was observed for seven studied compounds (Fig. 6). These results highlight the usefulness of the validated theoretical analysis of electronic properties prior to the practical synthesis of new acceptor materials, the design of which is based solely on the chemical intuition. Complex and mutually cancelling effects of the electron-withdrawing functional groups, saturation of the fullerene π system and the addition pattern type are difficult to predict empirically, whereas modern DFT theory allows for reliable, if not quantitative, data and trends to be revealed for these classes of molecules. | ||
| Fig. 6 Correlation of DFT calculated and experimental electron affinities for fullerenes studied in this work. A 3D model of the fullerene with the highest electron affinity, (10-1)(CN)2, is shown. | ||
Acknowledgements
We thank the U.S. NSF/CHE-1362302, NSF/CHE-1346572, National Institute of Health (grant R21CA140080), and the Colorado State University Research Foundation for partial financial support. The LT PES work was supported by the US Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences & Biosciences (X.–B.W.) and was performed at EMSL, a national scientific user facility sponsored by the US Department of Energy's Office of Biological and Environmental Research and located at PNNL. Popov acknowledges DFG (PO 1602/1-1) for financial support. The Research Computing Center of the Moscow State University is gratefully acknowledged for the computational facilities at the supercomputer “Chebyshev SKIF-MSU”.32References
- M. L. Tang and Z. A. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- Y. C. Chang, M. Y. Kuo, C. P. Chen, H. F. Lu and I. Chao, J. Phys. Chem. C, 2010, 114, 11595–11601 CAS.
- R. A. J. Janssen and J. Nelson, Adv. Mater., 2013, 25, 1847–1858 CrossRef CAS PubMed.
- D. C. Coffey, B. W. Larson, A. W. Hains, J. B. Whitaker, N. Kopidakis, O. V. Boltalina, S. H. Strauss and G. Rumbles, J. Phys. Chem. C, 2012, 116, 8916–8923 CAS.
- W. A. Braunecker, S. D. Oosterhout, Z. R. Owczarczyk, R. E. Larsen, B. W. Larson, D. S. Ginley, O. V. Boltalina, S. H. Strauss, N. Kopidakis and D. C. Olson, Macromolecules, 2013, 46, 3367–3375 CrossRef CAS.
- V. R. Koppolu, M. C. Gupta and L. Scudiero, Sol. Energy Mater. Sol. Cells, 2011, 95, 1111–1118 CrossRef CAS.
- Y. Abe, R. Hata and Y. Matsuo, Chem. Lett., 2013, 42, 1525–1527 CrossRef CAS.
- T. T. Clikeman, S. H. M. Deng, S. Avdoshenko, X.-B. Wang, A. A. Popov, S. H. Strauss and O. V. Boltalina, Chem. – Eur. J., 2013, 19, 15404–15409 CrossRef CAS PubMed.
- A. A. Popov, I. E. Kareev, N. B. Shustova, E. B. Stukalin, S. F. Lebedkin, K. Seppelt, S. H. Strauss, O. V. Boltalina and L. Dunsch, J. Am. Chem. Soc., 2007, 129, 11551–11568 CrossRef CAS PubMed.
- B. W. Larson, J. B. Whitaker, X. B. Wang, A. A. Popov, G. Rumbles, N. Kopidakis, S. H. Strauss and O. V. Boltalina, J. Phys. Chem. C, 2013, 117, 14958–14964 CAS.
- W. Han, H. Yoshida, N. Ueno and S. Kera, Appl. Phys. Lett., 2013, 103, 123303 CrossRef.
- H. Yoshida, Anal. Bioanal. Chem., 2014, 406, 2231–2237 CrossRef CAS PubMed.
- D.-L. Huang, P. D. Dau, H.-T. Liu and L.-S. Wang, J. Chem. Phys., 2014, 140, 224315 CrossRef PubMed.
- X.-B. Wang, H.-K. Woo, X. Huang, M. M. Kappes and L.-S. Wang, Phys. Rev. Lett., 2006, 96, 143002 CrossRef PubMed.
- M. Keshavarz-K, B. Knight, G. Srdanov and F. Wudl, J. Am. Chem. Soc., 1995, 117, 11371–11372 CrossRef CAS.
- G. Khairallah and J. B. Peel, J. Phys. Chem. A, 1997, 101, 6770–6774 CrossRef CAS.
- A. A. Popov, I. E. Kareev, N. B. Shustova, S. F. Lebedkin, S. H. Strauss, O. V. Boltalina and L. Dunsch, Chem. – Eur. J., 2008, 14, 107–121 CrossRef CAS PubMed.
- E. I. Dorozhkin, D. V. Ignat'eva, N. B. Tamm, A. A. Goryunkov, P. A. Khavrel, I. N. Ioffe, A. A. Popov, I. V. Kuvychko, A. V. Streletskiy, V. Y. Markov, J. Spandl, S. H. Strauss and O. V. Boltalina, Chem. – Eur. J., 2006, 12, 3876–3889 CrossRef CAS PubMed.
- K. P. Castro, Y. H. Jin, J. J. Rack, S. H. Strauss, O. V. Boltalina and A. A. Popov, J. Phys. Chem. Lett., 2013, 4, 2500–2507 CrossRef CAS.
- X. B. Wang and L. S. Wang, Rev. Sci. Instrum., 2008, 79, 073108 CrossRef PubMed.
- E. C. M. Chen and W. E. Wentworth, J. Chem. Phys., 1975, 63, 3183–3191 CrossRef CAS.
- D. Paolucci, F. Paolucci, M. Marcaccio, M. Carano and R. Taylor, Chem. Phys. Lett., 2004, 400, 389–393 CrossRef CAS.
- R. Meerheim, S. Olthof, M. Hermenau, S. Scholz, A. Petrich, N. Tessler, O. Solomeshch, B. Lüssem, M. Riede and K. Leo, J. Appl. Phys., 2011, 109, 103102 CrossRef.
- S. Sque, R. Jones, J. Goss, P. Briddon and S. Öberg, J. Phys.: Condens. Matter, 2005, 17, L21 CrossRef CAS.
- M. L. Tietze, L. Burtone, M. Riede, B. Lüssem and K. Leo, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 035320 CrossRef.
- J. Ristein, J. Phys. D: Appl. Phys., 2006, 39, R71 CrossRef CAS.
- A. G. Avent, K. Ala'a, B. W. Clare, D. L. Kepert, J. M. Street and R. Taylor, Org. Biomol. Chem., 2003, 1, 1026–1033 CAS.
- J. Holloway, E. Hope and J. Street, J. Chem. Soc., Perkin Trans. 2, 1998, 1845–1850 Search PubMed.
- I. V. Kuvychko, K. P. Castro, S. H. M. Deng, X.-B. Wang, S. H. Strauss and O. V. Boltalina, Angew. Chem., Int. Ed., 2013, 52, 4871–4874 CrossRef CAS PubMed.
- I. V. Kuvychko, S. N. Spisak, Y. S. Chen, A. A. Popov, M. A. Petrukhina, S. H. Strauss and O. V. Boltalina, Angew. Chem., Int. Ed., 2012, 51, 4939–4942 CrossRef CAS PubMed.
- O. V. Boltalina, L. N. Sidorov, A. Y. Borchshevskii, E. V. Sukhanova and E. V. Skokan, Rapid Commun. Mass Spectrom., 1993, 7, 1009–1011 CrossRef CAS.
- Vl. V. Voevodin, S. A. Zhumatiy, S. I. Sobolev, A. S. Antonov, P. A. Bryzgalov, D. A. Nikitenko, K. S. Stefanov and Vad. V. Voevodin, Practice of “Lomonosov” Supercomputer. Open Systems J., Open Systems Publ., Moscow, 2012, no. 7. (in Russian) Search PubMed.
- D. N. Laikov and Y. A. Ustynuk, Russ. Chem. Bull., 2005, 54, 820–826 CrossRef CAS.
- D. N. Laikov, Chem. Phys. Lett., 1997, 281, 151–156 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- T. T. Clikeman, I. V. Kuvychko, N. B. Shustova, Y.-S. Chen, A. A. Popov, O. V. Boltalina and S. H. Strauss, Chem. – Eur. J., 2013, 19, 5070 CrossRef CAS PubMed.
Footnote |
| † Synthesis of (10-1)(CN)2: The C70(CF3)10(CN)− anion was generated by adding an aliquot of a 6.41 mM ACN solution of NEt4CN (5.20 mL, 33.3 μmol CN−) to a yellow solution of 10-1 (25.7 mg, 16.8 μmol) in toluene (15 mL) at 23(2) °C (CN−:10-1 mol ratio = 2.00). The reaction mixture immediately became dark green. An aliquot of the solution (10.1 mL, 8.4 μmol C70(CF3)10(CN)−) was reacted with a 75 mM toluene solution of p-TsCN (5 mL, 376 μmol “CN+”, 45 equiv.) and the solution became yellow after 3 h. The solution was exposed to air, washed four times with water, and dried with MgSO4. The solvent was removed and the remaining solids were analyzed by 19F NMR and separated by HPLC: Cosmosil Buckyprep semi-preparative column, toluene/heptane = 60/40 mobile phase, 5 mL min−1, 300 nm detection, retention time = 3.2 minutes. Negative-ion Atmospheric Pressure Chemical Ionization mass spectrometry (NI-APCI-MS): calculated 1581.96, observed 1582.07 m/z. PES measurements: the spectroscopy and procedures used were described previously.20 Anions of C70 fullerene derivatives were generated by electrospraying a 0.1 mM acetonitrile solution of each fullerene mixed with TDAE donors. Anions generated were transported by RF ion guides into a cryogenic ion trap, where they were accumulated and cooled to 12 K, before being pulsed out at 10 Hz into an extraction zone of a time-of-flight (TOF) mass spectrometer. Mass-selected anions were first maximally decelerated and then intersected by 266 nm (4.661 eV) photons from a Nd:YAG laser in the photodetachment zone. The laser was operated at a 20 Hz repetition rate with ion beam off at alternating shots, enabling shot-by-shot background subtraction. Photoelectrons were collected at nearly 100% efficiency using the magnetic-bottle and analyzed in a 5.2 m long photoelectron flight tube. The TOF photoelectron spectrum was converted to the kinetic energy spectrum, calibrated by the known I− and ClO2− spectra. The binding energy spectrum presented in the paper was obtained by subtracting the kinetic energy spectrum from the photon energy used. The gas-phase EA of each compound was determined from the 0-0 transition in the 12 K LT-PES spectrum of each radical anion with an accuracy of 10 meV. DFT calculations: molecular structures of all studied species were optimized at the PBE/TZ2P level using Priroda code.33,34 Then, single point energy B3LYP35,36 calculations using the def2-TZVP basis set37 were performed using the Orca suite.38 |
| This journal is © the Owner Societies 2015 |