
Why is sulfuric acid a much stronger acid than ethanol? Determination of the contributions by inductive/field effects and electron-delocalization effects†
Kevin
Lynch
a,
Adam
Maloney
a,
Austin
Sowell
a,
Changwei
Wang
b,
Yirong
Mo
*b and
Joel M.
Karty
*a
aDepartment of Chemistry, Elon University, Elon, North Carolina, USA. E-mail: jkarty@elon.edu
bDepartment of Chemistry, Western Michigan University, Kalamazoo, Michigan, USA. E-mail: yirong.mo@wmich.edu
First published on 7th October 2014
Abstract
Two different and complementary computational methods were used to determine the contributions by inductive/field effects and by electron-delocalization effects toward the enhancement of the gas-phase deprotonation enthalpy of sulfuric acid over ethanol. Our alkylogue extrapolation method employed density functional theory calculations to determine the deprotonation enthalpy of the alkylogues of sulfuric acid, HOSO2–(CH2CH2)n–OH, and of ethanol, CH3CH2–(CH2CH2)n–OH. The inductive/field effect imparted by the HOSO2 group for a given alkylogue of sulfuric acid was taken to be the difference in deprotonation enthalpy between corresponding (i.e., same n) alkylogues of sulfuric acid and ethanol. Extrapolating the inductive/field effect values for the n = 1–6 alkylogues, we obtained a value of 51.0 ± 6.4 kcal mol−1 for the inductive/field effect for n = 0, sulfuric acid, leaving 15.4 kcal mol−1 as the contribution by electron-delocalization effects. Our block-localized wavefunction method was employed to calculate the deprotonation enthalpies of sulfuric acid and ethanol using the electron-localized acid and anion species, which were compared to the values calculated using the electron-delocalized species. The contribution from electron delocalization was thus determined to be 18.2 kcal mol−1, which is similar to the value obtained from the alkylogue extrapolation method. The two methods, therefore, unambiguously agree that both inductive/field effects and electron-delocalization effects have significant contributions to the enhancement of the deprotonation enthalpy of sulfuric acid compared with ethanol, and that the inductive/field effects are the dominant contributor.
Introduction
Sulfuric acid is one of the most important compounds worldwide, reflected by the fact that over 200 million tons of it are produced globally each year.1 In the United States, more sulfuric acid is produced than any other single chemical.2 Much of sulfuric acid's importance derives from the fact that it is a very strong acid, which is integral, for example, in the industrial synthesis of phosphoric acid for use in manufacturing fertilizers,3 as well as in the catalysis of a wide variety of chemical reactions, including the alkylation process in petroleum refining.4 Indeed, in aqueous solution, sulphuric acid is about 19 pKa units more acidic than ethanol.5,6 In the gas phase, in which there are no complications due to solvation, the experimentally measured deprotonation enthalpy of sulfuric acid is 309.6 ± 2.6 kcal mol−1,7 whereas that of ethanol is 377.4 ± 2.1 kcal mol−1 (ref. 8)—a difference of about 68 kcal mol−1.Despite its vast chemical importance, the origin of sulfuric acid's enhanced acid strength over alcohols is not well understood, in much the same way that the origin of the enhanced acid strength of carboxylic acids was not well understood until the turn of the twenty-first century. The couple decades prior to that saw a robust controversy9–30 surrounding the contributions by resonance and inductive/field effects to a carboxylic acid's enhanced acidity. The inductive/field effects arise from the electron-withdrawing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group attached directly to the reaction center, which serve to stabilize the negative charge that develops on the reaction-center oxygen in the carboxylate anion (Fig. 1a). Resonance, on the other hand, stabilizes that negative charge by allowing the charge to be shared over both oxygen atoms of the carboxylate anion (Fig. 1b, bottom), made possible by the conjugation of the orbitals of π symmetry from the OC–O− system (Fig. 1b, top).
O group attached directly to the reaction center, which serve to stabilize the negative charge that develops on the reaction-center oxygen in the carboxylate anion (Fig. 1a). Resonance, on the other hand, stabilizes that negative charge by allowing the charge to be shared over both oxygen atoms of the carboxylate anion (Fig. 1b, bottom), made possible by the conjugation of the orbitals of π symmetry from the OC–O− system (Fig. 1b, top).
 | ||
| Fig. 1 Inductive/field and resonance stabilization of a carboxylate anion. (a) The electron-withdrawing effect of the CO group decreases the concentration of negative charge on O−. (b) (top) Conjugation of the p orbitals in a carboxylate anion. (bottom) Resonance structures of a carboxylate anion, showing that the negative charge is shared over both oxygen atoms. | ||
Much like in carboxylic acids, the enhanced acid strength of sulfuric acid should have a major contribution from inductive/field effects. In the case of sulfuric acid, those inductive/field effects are provided by the HOSO2 group attached to the OH reaction center (Fig. 2a). The HOSO2 group is expected to be quite electron withdrawing inductively, similar to the methylsulfonyl group (CH3SO2),31 owing to the presence of the highly electronegative oxygen atoms and the moderately electronegative sulfur atom. Thus, the HOSO2 group should inductively stabilize the negative charge generated on the reaction-center oxygen in HSO4−.
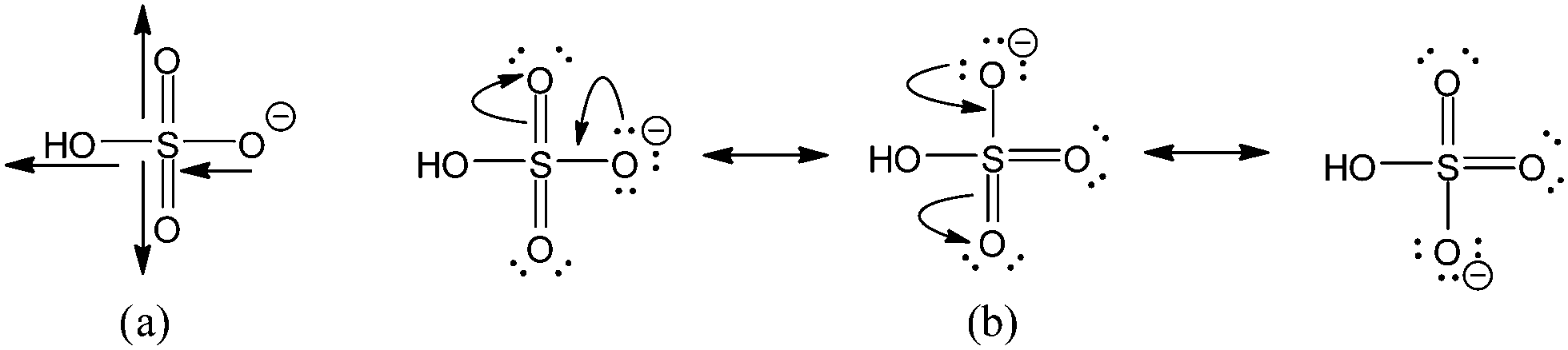 | ||
| Fig. 2 Inductive/field and resonance stabilization of HSO4−. (a) The electron-withdrawing effect of the HOSO2 group decreases the concentration of negative charge on O−. (b) Resonance structures of HSO4−, showing that the negative charge is shared over three oxygen atoms. | ||
It might also seem that resonance should have a significant contribution to the acid strength of sulfuric acid, given that resonance structures can be drawn to show the delocalization of the negative charge that develops in HSO4− (Fig. 2b). Drawing these resonance structures assumes that sulfur is hypervalent, where the π bond from each SO bond is constructed using a d orbital from sulfur. However, several recent studies have shown that the d orbitals from sulfur do not participate substantially in such π bonding,32–35 suggesting that this kind of resonance stabilization in HSO4− should be insignificant.
Nevertheless, there are other orbital interactions in HSO4− that can serve to delocalize the negative charge, thus leading to significant stabilization. These are the negative hyperconjugation interactions involving a lone pair of electrons from O− and the σ* orbitals of geminal S–O bonds (i.e., n → σ* interactions), as shown in Fig. 3.33
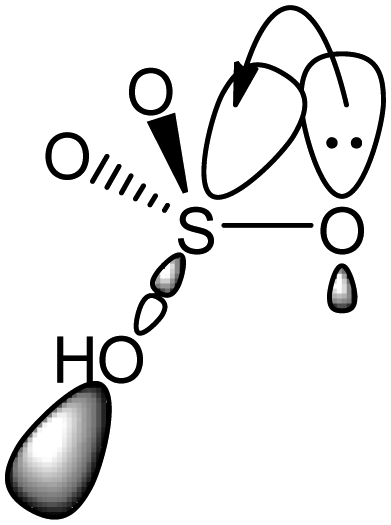 | ||
| Fig. 3 Negative hyperconjugation in HSO4−. A lone pair of electrons on O− is delocalized into the σ* orbital of an S–O bond. | ||
The question thus becomes, how much of sulfuric acid's enhancement in acid strength is attributed to these orbital interactions, and how much is attributed to inductive/field effects? Here in this study, we investigate this question using two different and complementary computational methods. One is the alkylogue extrapolation method, and the second is the block-localized wavefunction method. Both of these methods are described later.
To our knowledge, we are the first to provide an accurate evaluation of these kinds of contributions to the acid strength of sulfuric acid at the ab initio level. Denehy et al.,33 however, applied second-order perturbation analysis (as part of their natural bond order and natural resonance theory analyses) on HOSO3CH3 and HOSO3− to estimate the stabilization afforded each species by delocalizing interactions. They estimated 26.9 kcal mol−1 of stabilization in HOSO3CH3 and 93.7 kcal mol−1 of stabilization in HOSO3−. Assuming H2SO4 and HSO4− have similar stabilizations as HOSO3CH3 and HOSO3−, respectively, this would predict the orbital interactions to contribute about 67 kcal mol−1 to sulfuric acid's strength, which is essentially the entirety of the enhancement in deprotonation enthalpy of sulfuric acid over ethanol (68 kcal mol−1). As we show later, we believe this value to be greatly overestimated, by roughly a factor of 4.
Alkylogue extrapolation method
The alkylogue extrapolation method is based on the notion that saturated alkyl chains inserted between a reaction center and an attached group will disrupt the orbital interactions the reaction center has with the attached group. The alkylogues of sulfuric acid, in particular, are represented by Fig. 4.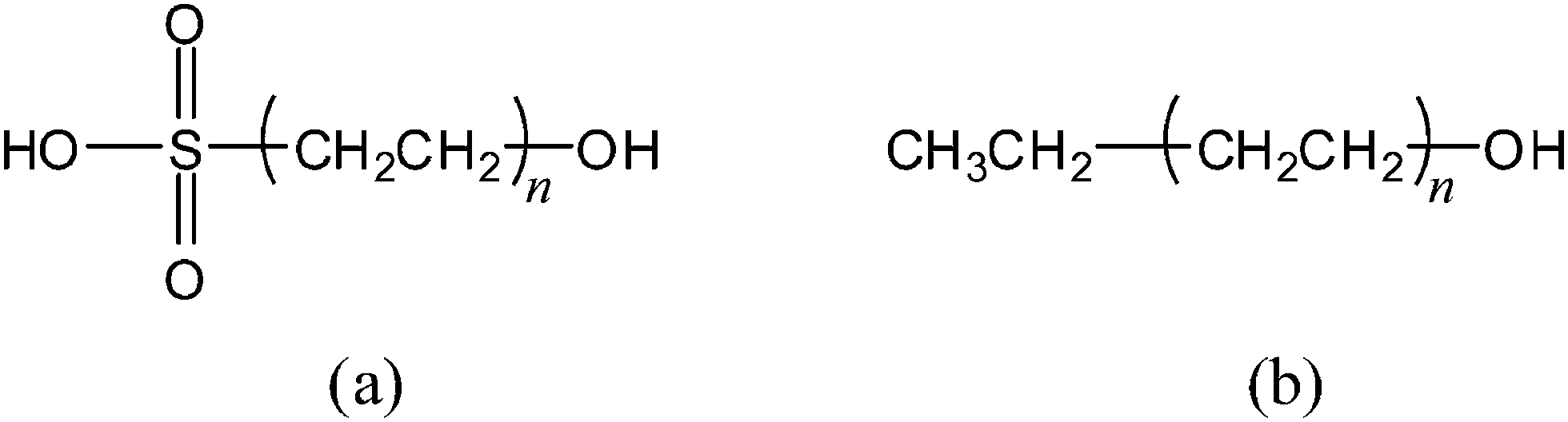 | ||
| Fig. 4 Alkylogues of sulfuric acid and ethanol. In each alkylogue of (a) sulfuric acid and (b) ethanol, an alkyl chain composed of n CH2CH2 groups is inserted between the OH reaction center and the HOSO2 and CH3CH2 groups, respectively. The parent H2SO4 and CH3CH2OH molecules are simply the respective n = 0 alkylogues. | ||
The difference in deprotonation enthalpy between a particular alkylogue of sulfuric acid and the corresponding (i.e., same value of n) alkylogue of ethanol, therefore, is attributed essentially entirely to inductive/field effects imposed by the HOSO2 group. After determining the inductive/field contributions for several different values of n, extrapolation of these values back to n = 0 yields the inductive/field effect contributed by the HOSO2 group in sulfuric acid. Finally, the component of sulfuric acid's enhancement in deprotonation enthalpy attributed to the direct orbital interactions between the reaction center and the HOSO2 group is determined by taking the difference between the extrapolated n = 0 inductive/field effect and the total enhancement in deprotonation enthalpy of sulfuric acid over ethanol.
The deprotonation enthalpy of each alkylogue of sulfuric acid should reflect some residual negative hyperconjugation between the reaction center and the bonds of the attached alkyl carbon. However, this effect should be essentially the same in the deprotonation enthalpy of the corresponding alkylogue of ethanol, so when the inductive/field effect is computed as described above, the impact that this residual negative hyperconjugation has should be effectively cancelled. Indeed, when this type of method was applied to para-substituted phenols,36 the results were in quite good agreement with the well-known Swain–Lupton resonance and inductive/field parameters.31
Block-localized wavefunction method
The block localized wavefunction (BLW) method is the simplest variant of ab initio valence bond (VB) theory.37–39 Unlike conventional molecular orbital (MO) theory, which is a top-down method with the use of MOs delocalized over the whole system, VB theory uses a bottom-up strategy and constructs wave functions with localized orbitals.40–43 While one Lewis structure typically describes a molecule rather well, the intramolecular electron delocalization (conjugation or hyperconjugation) is measured with the introduction of other less important and often ionic Lewis structures in terms of Pauling's resonance theory.In VB theory, a Lewis structure is defined by a Heitler–London–Slater–Pauling (HLSP) function,44 which can be expanded into 2N Slater determinants (N is the number of bonds). The basic idea of the BLW method is to reduce the number of Slater determinants for a VB function, thus dramatically reducing the computational costs. In the BLW method, we partition the molecular system into several blocks (or fragments or groups), and limit the block-localized MOs (BL-MOs) to expand within only one block and to be doubly occupied in closed-shell cases. In such a way, a HLSP function can be reduced to only one Slater determinant ΨL. BL-MOs in the same block are constrained to be orthogonal like in MO methods, but among different blocks they are non-orthogonal like in VB theory. If we allow all orbitals to expand in the whole space of primitive orbitals, the BLW will be upgraded to the Hartree–Fock (HF, or Kohn–Sham within the density functional theory, DFT) wave function ΨD, corresponding to a delocalized state, which is implicitly a superposition of all electron-localized states. Thus, the energy difference between ΨL and ΨD is defined as the electron delocalization energy (DE), according to eqn (1).
| DE = E(ΨD) − E(ΨL) | (1) |
Computational details
Alkylogue extrapolation method
All calculations were carried out assuming the gas phase, using the Gaussian 09 software package.45 Geometry optimizations and frequency calculations were performed on H2SO4, HSO4−, CH3CH2OH, CH3CH2O−, and each of their alkylogues through n = 6, all at the B3LYP/6-311++G(d,p) level of theory.46 The frequency calculations afforded the thermally corrected enthalpy of each species, and were also used to ensure that each optimized geometry had no imaginary frequencies, thus making it a true local energy minimum. Deprotonation enthalpies were computed by subtracting the calculated enthalpy of the acid from that of its corresponding anion, followed by adjustment for the production of a bare proton.For each alkylogue, the alkyl chain was in the all-anti conformation. This ensures that the torsional strain in each alkyl chain is minimized and that artifacts imposed by the addition of an alkyl chain are consistent from one alkylogue to another.
Block-localized wavefunction method
The BLW method was applied to H2SO4 (or HOSO2–OH) and HSO4− (or HOSO2–O−), as well as C2H5–OH and C2H5–O−, using the 6-31+G(d,p) basis set. Geometries were optimized at the B3LYP/6-311++G(d,p) level. To derive the BLW for the electron-localized state without the electron delocalization between the deprotonation-related OH group in H2SO4/C2H5OH or O− group in HSO4−/C2H5O− and the remaining fragment (HSO3 or C2H5), we defined three blocks for all systems. One block was for the HOSO2 or C2H5 fragment with 40 or 16 electrons, respectively, one block was for the OH or O− group with 8 electrons, and the last block was for the S–O or C–O single bond connecting the HOSO2 or C2H5 and OH/O− fragments with 2 electrons. The BLW calculations were carried out using the Xiamen Valence Bond (XMVB) program.47Results
Alkylogue extrapolation method
The calculated deprotonation enthalpies (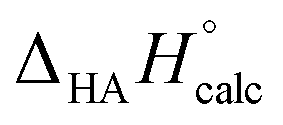 ) of sulfuric acid, ethanol, and their n = 1–6 alkylogues are presented in Table 1. The calculated value of the inductive/field effect for each alkylogue of sulfuric acid, denoted as Ind(n), is computed by subtracting the deprotonation enthalpy of the nth alkylogue of sulfuric acid from the deprotonation enthalpy of the corresponding (i.e., same n) alkylogue of ethanol. Also included in the table is the calculated distance between S and O−, r(S–O−), in each geometry-optimized alkylogue of HSO4−, because those values were used for extrapolation of the Ind(n) values. The value for Ind(0) appears in parentheses because it was obtained by extrapolating the n = 1–6 values.
) of sulfuric acid, ethanol, and their n = 1–6 alkylogues are presented in Table 1. The calculated value of the inductive/field effect for each alkylogue of sulfuric acid, denoted as Ind(n), is computed by subtracting the deprotonation enthalpy of the nth alkylogue of sulfuric acid from the deprotonation enthalpy of the corresponding (i.e., same n) alkylogue of ethanol. Also included in the table is the calculated distance between S and O−, r(S–O−), in each geometry-optimized alkylogue of HSO4−, because those values were used for extrapolation of the Ind(n) values. The value for Ind(0) appears in parentheses because it was obtained by extrapolating the n = 1–6 values.
| n | H2SO4 | CH3CH2OH | ||||
|---|---|---|---|---|---|---|
| r (S–O−) (Å) |
|
|
|
|
Ind(n)d | |
| a Thermally corrected values calculated at the B3LYP/6-311++G(d,p) level of theory. b Experimental values not found in the NIST database (ref. 48) are indicated by a dash. c Calculated distance between S and O− from the geometry-optimized alkylogues of HSO4−. d Each value of the inductive/field effect is computed by subtracting the deprotonation enthalpy of the nth alkylogue of sulfuric acid from the deprotonation enthalpy of the corresponding (i.e., same n) alkylogue of ethanol. e See ref. 7; gas-phase equilibrium measurement. f See ref. 8; gas-phase equilibrium measurement. g Numbers in parentheses are derived from the extrapolation of the n = 1–6 values. h See ref. 49; gas-phase equilibrium measurement. i See ref. 50; kinetic method. | ||||||
| 0 | 1.47 | 308.1 | 309.6 ± 2.6e | 374.5 | 377.4 ± 2.1f | (51.0)g |
| 1 | 4.04 | 352.9 | — | 373.9 | 375.4 ± 2.1f | 21.0 |
| 2 | 6.57 | 363.8 | — | 373.5 | 374.0 ± 2.1h | 9.7 |
| 3 | 9.12 | 368.5 | — | 373.4 | 374.3 ± 2.1h | 4.9 |
| 4 | 11.68 | 370.5 | — | 373.4 | 372.9 ± 2.0i | 2.8 |
| 5 | 14.24 | 371.5 | — | 373.3 | — | 1.8 |
| 6 | 16.80 | 371.9 | — | 373.3 | — | 1.4 |




Fig. 5 is the plot of Ind(n) against the S–O− distance in each geometry-optimized alkylogue of HSO4−. The dashed curve represents the function derived from the nonlinear least-squares fit of the data, as explained later in the Discussion section.
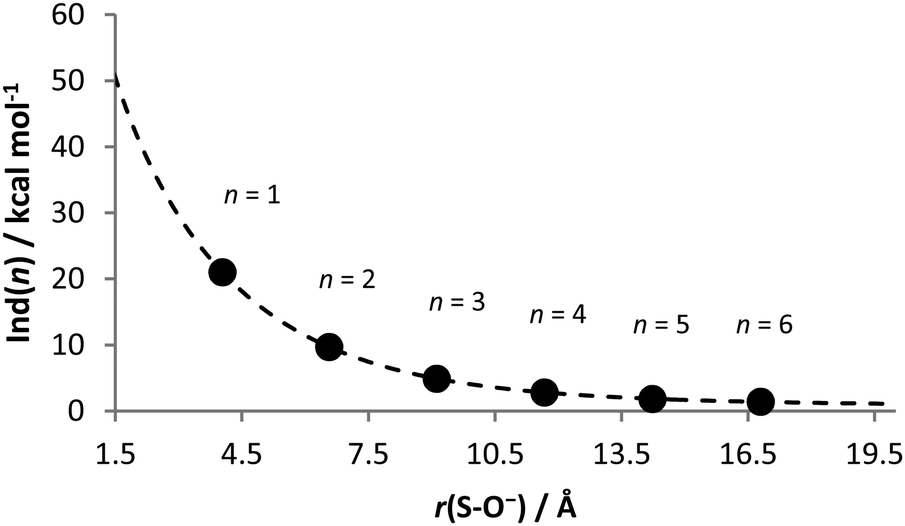 | ||
| Fig. 5 Extrapolation of the inductive/field effects. The data points represent the contribution by inductive/field effects toward the deprotonation-enthalpy enhancement in each alkylogue of H2SO4. The dashed curve represents the nonlinear least-squares fit of those data. | ||
Block-localized wavefunction method
The above alkylogue extrapolation method derives the inductive/field contribution to the deprotonation-enthalpy enhancement of sulfuric acid compared to ethanol, and the electron delocalization contribution is indirectly obtained by subtracting the inductive/field contribution from the difference in deprotonation enthalpy between sulfuric acid and ethanol. In contrast, the BLW method is intended to derive the electron delocalization contribution directly. Table 2 summarizes the BLW computation results. The difference between the electron delocalization energies in sulfuric acid and its deprotonated product is 40.7 kcal mol−1, which corresponds to the electron delocalization contribution to the deprotonation of sulfuric acid. However, electron delocalization effects exist in ethanol and its deprotonated anion as well. The difference between delocalization energies of these two species yields 22.5 kcal mol−1 as the contribution by electron delocalization to the deprotonation enthalpy of ethanol. Comparing the values for both sulfuric acid and ethanol, we estimate that electron delocalization contributes 18.2 kcal mol−1 to the acidity enhancement of sulfuric acid with reference to ethanol, while the inductive/field effect is responsible for the remaining 48.2 kcal mol−1.| E(ΨD) | E(ΨL) | DE | |
|---|---|---|---|
| H2SO4 | −698.05189 | −698.01769 | −21.5 |
| HSO4− | −697.54499 | −697.44582 | −62.2 |
| C2H5OH | −154.09492 | −154.07400 | −13.1 |
| C2H5O− | −153.46222 | −153.40541 | −35.6 |
Discussion
Alkylogue extrapolation method
The B3LYP/6-311++G(d,p) level was chosen for these calculations because it reproduces well the difference in the experimental gas-phase deprotonation enthalpies between sulfuric acid and ethanol, while not being computationally very expensive. This is particularly important for the large alkylogues, which have up to 17 heavy atoms and a large number of degrees of freedom. For each deprotonation enthalpy, multiple experimental values are available,48 derived from multiple different techniques. To ensure the best accuracy for the relative numbers (which is what the alkylogue extrapolation method depends upon), the experimental numbers used for comparison (Table 1) are the ones derived from equilibrium measurements, yielding a difference of 67.8 kcal mol−1. The B3LYP/6-311++G(d,p) calculated difference in deprotonation enthalpies is 66.4 kcal mol−1. To establish that our results are not significantly dependent on the choice of the exchange–correlation functional, we also calculated the deprotonation enthalpies of sulphuric acid and ethanol at the mPW1PW91/6-311++G(d,p) level of theory.51 At that level of theory, the deprotonation of sulphuric acid is 67.2 kcal mol−1 more exothermic than ethanol, which is nearly equal to the value of 66.4 kcal mol−1 reported above.Experimental deprotonation enthalpies for the n = 1–4 alkylogues of ethanol are also available for comparison (Table 1). The experimental values for n = 1–3 are all derived from gas-phase equilibrium measurements. They all lie within a 1.1 kcal mol−1 range, with the value for n = 1 being the largest. Similarly, the calculated values for n = 1–3 all lie within a range of 0.5 kcal mol−1, with the value for n = 1 being the largest. The experimental value for n = 4 appears to be anomalously low, probably because it was derived from a kinetic method rather than equilibrium measurements.
The calculated deprotonation enthalpies of the ethanol alkylogues decrease monotonically from n = 0–6, owing to the increasingly polarizable alkyl chain. This is explained by the fact that with greater polarizability, the negative charge that is produced in the alkoxide anion enjoys greater internal solvation.52 A saturation effect is exhibited by the leveling off of these values around 373.3 kcal mol−1.
The experimental deprotonation enthalpies for the n = 0–3 alkylogues, on the other hand, exhibit a minimum at n = 2. As Higgins and Bartmess49 point out, this is due to the ability of the alkyl chain to bend, allowing for more effective internal solvation when the chain becomes long enough. Such an effect is not reflected by our calculated numbers because all of the calculated geometries have an extended, all-anti alkyl chain. In fact, for the purpose of our alkylogue method, the geometries with the extended alkyl chains are preferable because, when computing the difference in deprotonation enthalpies between corresponding alkylogues of sulfuric acid and ethanol, artifacts due to differences in this internal solvation are avoided.
Unlike what is observed for the alkylogues of ethanol, the calculated deprotonation enthalpies of the n = 1–6 alkylogues of sulfuric acid increase monotonically. This is because the SO3H group is becoming increasingly distant from the reaction center, so the charge stabilization in the conjugate base is decreasing. More specifically, as shown in Table 1, this is due to the decreasing stabilization by the inductive/field effects, Ind(n).
The contribution by inductive/field effects to the deprotonation enthalpy of sulfuric acid—that is, Ind(0)—has a lower bound of about 21.0 kcal mol−1, which is the contribution to the n = 1 alkylogue. To obtain a more precise number, the values of Ind(n) were plotted against r(S–O−) of the geometry-optimized alkylogues of HSO4−, as shown in Fig. 5. The dependent variable used was r(S–O−) because the stabilization of the negative charge is expected to diminish as the distance between O− and the HOSO2 group increases. A nonlinear least-squares fit of these data allowed for the extrapolation of Ind(n) back to n = 0 to yield 51.0 ± 6.4 kcal mol−1 as the contribution by inductive/field effects toward the deprotonation-enthalpy enhancement of sulfuric acid over ethanol.
The general function that was used for the nonlinear least-squares fit is provided in eqn (2).
| Ind(n) = [Ae−br(S–O−)] + C/([r(S–O−)]d) | (2) |
Here, A, b, C and d are fitting parameters. The first term on the right side of eqn (2) is an exponential decay, which is expected for inductive effects.53,54 The second term on the right side of the equation is an inverse-power relationship, which is the general form expected for through-space intermolecular interactions.55 Least-squares fitting of this function to the Ind(n) data yields: A = 69.09 ± 2.36; b = 0.35 ± 0.01; C = 13.40 ± 4.73; d = 0.86 ± 0.22. The resulting equation reproduces the Ind(n) data with an average discrepancy of 0.02 kcal mol−1. To extrapolate Ind(n) to n = 0, the value of 1.47 Å was substituted for r(S–O−), according to Table 1, to yield 51.0 ± 6.4 kcal mol−1 as the contribution by inductive/field effects toward the deprotonation-enthalpy enhancement of sulfuric acid over ethanol. The 6.4 kcal mol−1 uncertainty reflects the standard deviations of A, b, C and d obtained from the nonlinear fit.
The difference in the calculated deprotonation enthalpies between sulfuric acid and ethanol, 66.4 kcal mol−1, is taken to be the sum of the inductive/field effect and the delocalization effect from direct orbital interactions. Thus, the delocalization effect is worth approximately 15.4 kcal mol−1.
Block-localized wavefunction method
The BLW computations use three blocks to measure the energetic contribution by conjugation/hyperconjugation between the –OH/O− group and the HO–SO2 or C2H5 group. The delocalization energy (DE) of each anion (Table 2) is significantly greater than the DE of its corresponding acid. This is consistent with a greater extent of electron redistribution for each anion upon going from the electron-localized structure to the delocalized structure, as exemplified by the electron-density difference maps calculated for H2SO4 and HSO4− (Fig. 6). | ||
| Fig. 6 Electron density difference (EDD) maps for the intramolecular electron delocalization in H2SO4 and HSO4−. (a) EDD map for H2SO4. (b) EDD for HSO4−. The red color means an increase in electron density upon going from the localized structure to the delocalized structure, whereas the blue color represents a decrease in electron density. The red and blue regions are larger for HSO4−, indicating a greater extent of electron redistribution. | ||
The difference between the electron delocalization energies in H2SO4 and HSO4− is 40.7 kcal mol−1, but the corresponding difference in C2H5OH and C2H5O− is also significant, at 22.5 kcal mol−1. Thus, overall the electron delocalization effect contributes 18.2 kcal mol−1 to the deprotonation-enthalpy enhancement of sulfuric acid compared with ethanol. This value is close to the value of 15.4 kcal mol−1 indirectly derived from the alkylogue extrapolation method. These two very different approaches, therefore, consistently suggest that the intramolecular electron delocalization effect makes an important contribution to the strong acidity of sulfuric acid.
Conclusions
We carried out two distinct and complementary computational methods to determine the contributions by inductive/field effects and by electron-delocalization effects toward the enhancement of the gas-phase deprotonation enthalpy of sulfuric acid over ethanol. One was the alkylogue extrapolation method, and the other was the block-localized wavefunction method. In our alkylogue extrapolation method, we used density functional theory to calculate the deprotonation enthalpy of the alkylogues of sulfuric acid, HOSO2–(CH2CH2)n–OH, and of ethanol, CH3CH2–(CH2CH2)n–OH. The difference in deprotonation enthalpy between corresponding (i.e., same n) alkylogues of sulfuric acid and ethanol was taken to be the inductive/field effect imparted by the HOSO2 group in HOSO2–(CH2CH2)n–OH. The inductive/field effect values for the n = 1–6 alkylogues were extrapolated to n = 0 to yield 51.0 ± 6.4 kcal mol−1 for the inductive/field effect in sulfuric acid. Subtracting this value from the difference in the calculated deprotonation enthalpy between sulfuric acid and ethanol, we obtain 15.4 kcal mol−1 for the contribution by electron-delocalization effects. In our block-localized wavefunction method, the deprotonation enthalpy was calculated for sulfuric acid using the electron-delocalized acid and anion, and again using the electron-localized species. The same was done for the corresponding ethanol species. This yields 18.2 kcal mol−1 as the contribution by electron delocalization toward sulfuric acid's enhancement in deprotonation enthalpy, leaving 48.2 kcal mol−1 as the contribution by inductive/field effects. The two different methods therefore agree that both inductive/field effects and electron-delocalization effects have significant contributions to the enhanced deprotonation enthalpy of sulfuric acid over ethanol, and that inductive/field effects have a substantially greater contribution.Acknowledgements
JK acknowledges financial support from a grant by the U.S. National Science Foundation, CHE-105883. YM acknowledges the support of the U.S. National Science Foundation under Grants CHE-1055310 and CNS-1126438.Notes and references
- The Essential Chemical Industry Online: Sulfuric Acid, http://www.essentialchemicalindustry.org/chemicals/sulfuric-acid.html, accessed July 21, 2014.
- Acid Plant New Source Review Enforcement Initiative, http://www2.epa.gov/enforcement/acid-plant-new-source-review-enforcement-initiative, accessed July 21, 2014.
- P. Becker, Phosphates and Phosphoric Acid: Raw Materials: Technology, and Economics of the Wet Process, Marcel Dekker, Inc., 1989 Search PubMed.
- J. H. Gary, G. E. Handwerk and M. J. Kaiser, Petroleum Refining: Technology and Economics, Marcel Dekker, Inc., 2005 Search PubMed.
- M. Kalthoff, Treatise on Analytical Chemistry, Interscience Encyclopedia, Inc., 1959 Search PubMed.
- P. Ballinger, J. Am. Chem. Soc., 1960, 82, 795 CrossRef CAS.
- A. A. Viggiano, M. J. Henchman, F. Dale, C. A. Deakyne and J. F. Paulson, J. Am. Chem. Soc., 1992, 114, 4299 CrossRef CAS.
- J. E. Bartmess, J. A. Scott and R. T. McIver, Jr., J. Am. Chem. Soc., 1979, 101, 6047 Search PubMed.
- G. W. Wheland, Resonance in Organic Chemistry, Wiley, 1955 Search PubMed.
- T. D. Thomas, J. Chem. Soc., Perkin Trans. 2, 1994, 1945 RSC.
- J. March, Advanced Organic Chemistry, Wiley, 1985 Search PubMed.
- T. H. Lowry and K. S. Richardson, Mechanism and Theory in Organic Chemistry, HarperCollins, 1987 Search PubMed.
- M. R. F. Siggel and T. D. Thomas, J. Am. Chem. Soc., 1986, 108, 4360 CrossRef CAS.
- O. Exner, J. Org. Chem., 1988, 53, 1810 CrossRef CAS.
- O. Exner and P. Carsky, J. Am. Chem. Soc., 2001, 123, 9564 CrossRef CAS PubMed.
- M. J. S. Dewar and K. L. Krull, J. Chem. Soc., Chem. Commun., 1990, 4, 333 RSC.
- M. Godfrey, Tetrahedron Lett., 1990, 31, 5181 CrossRef CAS.
- R. W. Taft, I. A. Koppel, R. D. Topsom and F. Anvia, J. Am. Chem. Soc., 1990, 112, 2047 CrossRef CAS.
- C. L. Perrin, J. Am. Chem. Soc., 1991, 113, 2865 CrossRef CAS.
- F. G. Bordwell and A. V. Satish, J. Am. Chem. Soc., 1994, 116, 8885 CrossRef CAS.
- P. C. Hiberty and C. P. Byrman, J. Am. Chem. Soc., 1995, 117, 9875 CrossRef CAS.
- J. D. M. Neto and M. A. C. Nascimento, J. Phys. Chem., 1996, 100, 15105 CrossRef.
- M. R. F. Siggel, A. R. Streitwieser, Jr. and T. D. Thomas, J. Am. Chem. Soc., 1988, 110, 8022 CrossRef CAS.
- T. D. Thomas, T. X. Carroll and M. R. F. Siggel, J. Org. Chem., 1988, 53, 1812 CrossRef CAS.
- T. D. Thomas, M. R. F. Siggel and A. Streitwieser, THEOCHEM, 1988, 165, 309 CrossRef.
- K. B. Wiberg and K. E. Laidig, J. Am. Chem. Soc., 1988, 110, 1872 CrossRef CAS.
- K. B. Wiberg, J. Am. Chem. Soc., 1990, 112, 3379 CrossRef CAS.
- D. Ji and T. D. Thomas, J. Phys. Chem., 1994, 98, 4301 CrossRef CAS.
- K. B. Wiberg, J. Ochterski and A. Streitwieser, J. Am. Chem. Soc., 1996, 118, 8291 CrossRef CAS.
- P. R. Rablen, J. Am. Chem. Soc., 2000, 122, 357 CrossRef CAS.
- C. G. Swain and E. C. Lupton, J. Am. Chem. Soc., 1968, 90, 4328 CrossRef CAS.
- J.-F. Boily, J. Phys. Chem. A, 2002, 106, 4718 CrossRef CAS.
- E. Denehy, J. M. White and S. J. Williams, Inorg. Chem., 2007, 46, 8871 CrossRef CAS PubMed.
- A. E. Reed and P. v. R. Schleyer, J. Am. Chem. Soc., 1990, 112, 1434 CrossRef CAS.
- D. B. Chesnut and L. D. Quin, J. Comput. Chem., 2004, 25, 734 CrossRef CAS PubMed.
- J. B. Barbour and J. M. Karty, J. Phys. Org. Chem., 2005, 18, 210 CrossRef CAS.
- Y. Mo, Nat. Chem., 2010, 2, 666 CrossRef CAS PubMed.
- Y. Mo and S. D. Peyerimhoff, J. Chem. Phys., 1998, 109, 1687 CrossRef CAS PubMed.
- Y. Mo, L. Song and Y. Lin, J. Phys. Chem. A, 2007, 111, 8291 CrossRef CAS PubMed.
- Valence Bond Theory, ed. D. L. Cooper, Amsterdam, 2002 Search PubMed.
- G. A. Gallup, Valence Bond Methods: Theory and Applications, Cambridge University Press, 2002 Search PubMed.
- S. S. Shaik and P. C. Hiberty, A Chemist's Guide to Valence Bond Theory, Wiley, 2008 Search PubMed.
- W. Wu, P. Su, S. Shaik and P. C. Hiberty, Chem. Rev., 2011, 111, 7557 CrossRef CAS PubMed.
- L. C. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, Cornell University Press, 1960 Search PubMed.
- M. J. Frisch, et al., Gaussian 09.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- L. Song, Z. Chen, F. Ying, J. Song, X. Chen, P. Su, Y. Mo, Q. Zhang and W. Wu, XMVB 2.0: An ab initio Non-Orthogonal Valence Bond Program, Xiamen University, Xiamen 361005, China, 2012 Search PubMed.
- J. E. Bartmess, in NIST Chemistry WebBook, NIST Standard Reference Database 69, ed. W. G. Mallard and P. J. Linstrom, National Institute of Standards and Technology, Gaithersburg, MD, 1998 Search PubMed.
- P. R. Higgins and J. E. Bartmess, Int. J. Mass Spectrom., 1998, 175, 71 CrossRef CAS.
- M. J. Haas and A. G. Harrison, Int. J. Mass Spectrom. Ion Processes, 1993, 124, 115 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664 CrossRef CAS PubMed.
- J. I. Brauman and L. K. Blair, J. Am. Chem. Soc., 1970, 92, 5986 CrossRef CAS.
- P. E. Peterson and C. Casey, Tetrahedron Lett., 1963, 4, 1569 CrossRef.
- G. Bianchi, O. W. Howarth, C. J. Samuel and G. Vlahov, J. Chem. Soc., Perkin Trans. 2, 1995, 1427 RSC.
- S. Tsuzuki, in Interactions with Aromatic Rings, ed. D. J. Wales, New York, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates, absolute energies, thermally corrected enthalpies, and number of imaginary frequencies for each calculated structure; complete citation for ref. 42. See DOI: 10.1039/c4cp04110k |
| This journal is © the Owner Societies 2015 |