
Separating mixtures by exploiting molecular packing effects in microporous materials†
Rajamani
Krishna
Van ‘t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: r.krishna@contact.uva.nl; Fax: +31 20 525 5604; Tel: +31 20 627 0990
First published on 7th November 2014
Abstract
We examine mixture separations with microporous adsorbents such as zeolites, metal–organic frameworks (MOFs) and zeolitic imidazolate frameworks (ZIFs), operating under conditions close to pore saturation. Pore saturation is realized, for example, when separating bulk liquid phase mixtures of polar compounds such as water, alcohols and ketones. For the operating conditions used in industrial practice, pore saturation is also attained in separations of hydrocarbon mixtures such as xylene isomers and hexane isomers. Separations under pore saturation conditions are strongly influenced by differences in the saturation capacities of the constituent species; the adsorption is often in favor of the component with the higher saturation capacity. Effective separations are achieved by exploiting differences in the efficiency with which molecules pack within the ordered crystalline porous materials. For mixtures of chain alcohols, the shorter alcohol can be preferentially adsorbed because of its higher saturation capacity. With hydrophilic adsorbents, water can be selectively adsorbed from water–alcohol mixtures. For separations of o-xylene–m-xylene–p-xylene mixtures, the pore dimensions of MOFs can be tailored in such a manner as to allow optimal packing of the isomer that needs to be adsorbed preferentially. Subtle configurational differences between linear and branched alkane isomers result in significantly different packing efficiencies within the pore topology of MFI, AFI, ATS, and CFI zeolites. A common characteristic feature of most separations that are reliant on molecular packing effects is that adsorption and intra-crystalline diffusion are synergistic; this enhances the separation efficiencies in fixed bed adsorbers.
 Rajamani Krishna | Rajamani Krishna is a Professor at the University of Amsterdam in the Netherlands. His major research focus is on adsorption and diffusion in nanoporous crystalline materials. He has published three text books, more than 400 peer-reviewed journal articles, and holds several patents. A complete list of his research contributions can be found on Google Scholar: http://scholar.google.nl/citations?user=cKqtQ0MAAAAJ&hl=en. According to the latest statistics on Google scholar, his publications have been cited a total of 21 |
1. Introduction
Microporous materials such as zeolites, metal–organic frameworks (MOFs), and zeolitic imidazolate frameworks (ZIFs) offer considerable potential as energy-efficient alternatives to conventional separation processes such as distillation, absorption, and extraction.The majority of current research on MOFs and ZIFs appears to be focused on separations of mixtures of light gaseous compounds. A common feature of separations of gaseous mixtures is that even for operations at pressures up to 1 MPa, pore saturation conditions are often not reached; these separations are largely reliant on differences in adsorption strengths and binding energies. For example, the selective adsorption of CO2 from mixtures containing N2, H2, CO, and CH4 can be realized by selective binding of CO2 with either the metal atoms (M) of CuBTC (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cu3(BTC)2 with BTC = 1,3,5-benzenetricarboxylate, also known as HKUST-1),1 Cu-TDPAT,2 M-MOF-74 (Mg2(dobdc))3,4 or the extra-framework cations of NaX zeolite.5 The quadrupole moment of N2 is about four times that of O2; as a consequence, for zeolites LiX, LTA-4A, and LTA-5A, the adsorption selectivity is in favor of N2.6 The differences in the polarizabilities of noble gases such as Kr and Xe can be exploited to recover each of these components in adsorbers packed with NiMOF-747,8 or CuBTC.7,9
Cu3(BTC)2 with BTC = 1,3,5-benzenetricarboxylate, also known as HKUST-1),1 Cu-TDPAT,2 M-MOF-74 (Mg2(dobdc))3,4 or the extra-framework cations of NaX zeolite.5 The quadrupole moment of N2 is about four times that of O2; as a consequence, for zeolites LiX, LTA-4A, and LTA-5A, the adsorption selectivity is in favor of N2.6 The differences in the polarizabilities of noble gases such as Kr and Xe can be exploited to recover each of these components in adsorbers packed with NiMOF-747,8 or CuBTC.7,9
For mixture adsorption, let us define the fractional occupancy within the pores, θt
 | (1) |
Fig. 1a shows the variation of θt with total gas pressure, pt, for adsorption of 20/80 CO2/H2 mixtures in Cu-TDPAT, 50/50 CO2/CH4 mixtures in NiMOF-74, and 15/85 CO2/N2 mixtures in MgMOF-74. For all these separations, under the range of operating conditions used in practice (indicated by the shaded areas), the fractional pore occupancy θt is commonly below 0.6. The separation characteristics for such mixtures are dictated by differences in one or more of the following characteristics: (a) van der Waals interactions and polarizability, (b) electrostatic interactions, and (c) π-electron transfers.
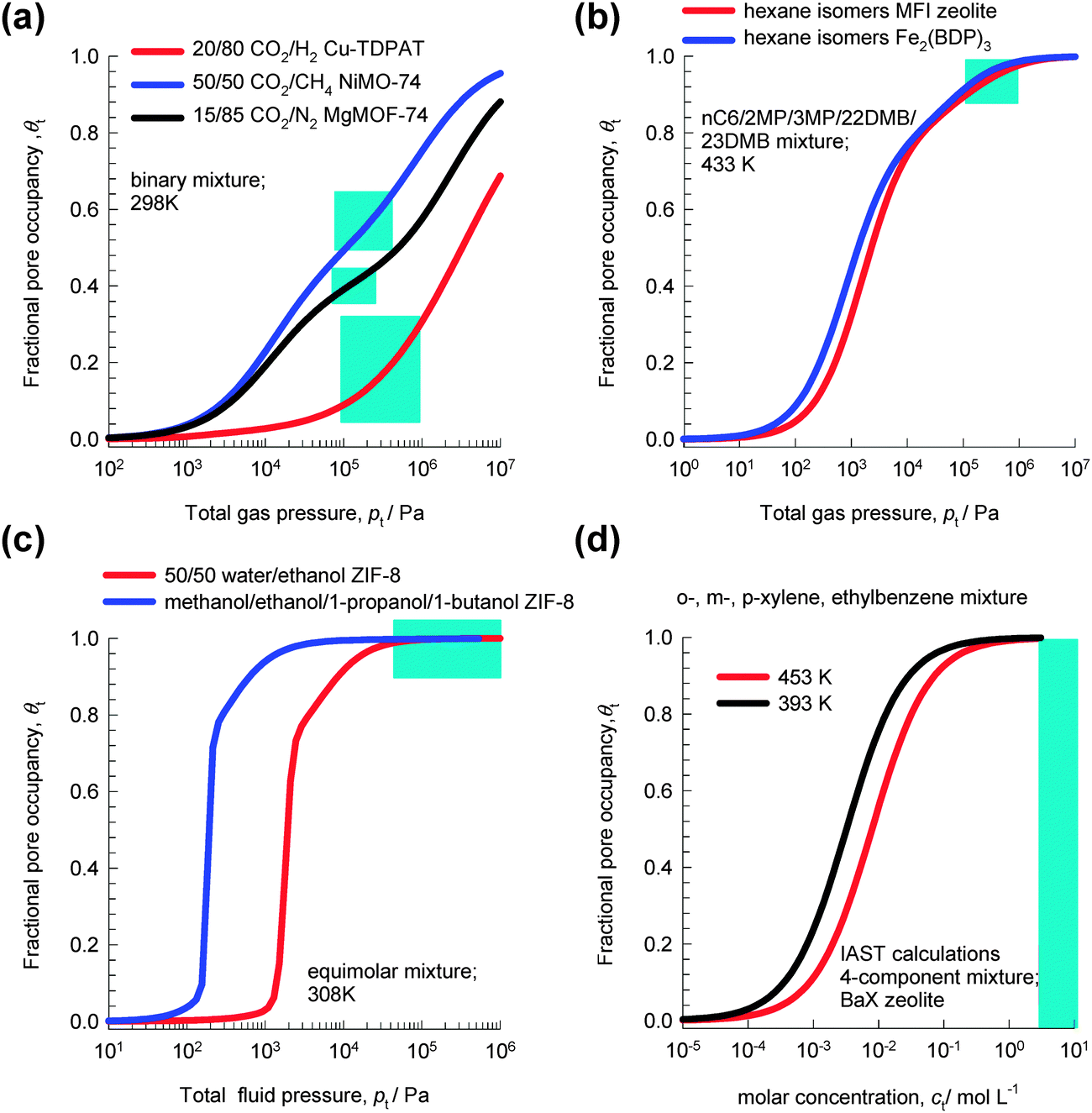 | ||
| Fig. 1 The variation of the fractional pore occupancy, θt, as a function of the bulk fluid phase pressure, pt, for adsorption of (a) 20/80 CO2/H2 mixtures in Cu-TDPAT, 50/50 CO2/CH4 mixtures in NiMOF-74, and 15/85 CO2/N2 mixtures in MgMOF-74, (b) nC6/2MP/3MP/22DMB/23DMB mixtures in MFI and Fe2(BDP)3, (c) equimolar water–ethanol and methanol–ethanol–1-propanol–1-butanol mixtures in ZIF-8, (d) o-xylene–m-xylene–p-xylene–ethylbenzene mixtures in BaX zeolite. See ESI† for further calculation details. | ||
For separation of mixtures of hexane isomers, industrial processes often operate under conditions in which pore saturation prevails, i.e. θt ≈ 1; see calculations in Fig. 1b for separations with Fe2(BDP)3 and MFI zeolite.10
Fig. 1c shows the variation of θt for adsorption of equimolar water–ethanol and methanol–ethanol–1-propanol–1-butanol mixtures in ZIF-8. We note that for pressures higher than 100 kPa, the pores are saturated. Typically, separations of dilute solutions of bioalcohols involve liquid phase mixtures under near ambient conditions;11 this ensures pore saturation.
Industrial separations of o-xylene–m-xylene–p-xylene–ethylbenzene mixtures with BaX zeolite are currently carried out in simulated moving bed (SMB) adsorbers in which the bulk phase is in the liquid state, typically with molar densities of 7–10 mol L−1.12,13 From the adsorption equilibrium data in Fig. 1d, we see that pore saturation conditions prevail in industrial separations.
In contrast to separations under conditions for which θt < 0.6, the separations under conditions close to pore saturation (i.e. θt ≈ 1) are significantly influenced by differences in saturation capacities, that reflect differences in the efficiencies by which the molecules pack, or stack, themselves within the microporous channels. The elucidation, and elaboration, of such molecular packing, or entropy, effects that favor adsorption of smaller molecules is the primary objective of this article. By a careful, detailed, analysis of a wide variety of separations we shall highlight a variety of separation strategies that can be employed in practice in order to increase the efficacies of separations. Often, such strategies are not intuitively obvious. The separations that are examined in this article include (a) mixtures of alcohols, (b) water–alcohol mixtures, (c) water–hydrocarbons, (d) C4 hydrocarbons, (e) hexane isomers, (f) xylene isomers, (g) chlorofluorocarbons, and (h) ethylbenzene–styrene mixtures. We shall demonstrate that molecular packing effects often overshadow the influences of adsorption strengths and binding energies, of paramount importance in separations at low pore occupancies, θt.
The secondary objective of this Perspective is to demonstrate the possibility of tailoring the pore geometries in order to enhance the separation selectivity in favor of one of the components. The use of bespoke-tailored MOFs for selective adsorption of p-xylene from a mixture of xylene isomers may result in significant improvements in current technologies that use BaX zeolite.
The ESI† accompanying our article provides (a) structural information (unit cell dimension, pore volumes, framework density, cage volumes, channel dimensions, pore landscapes) on zeolites, MOFs, and ZIFs, (b) details of configurational-bias Monte Carlo (CBMC) simulation methodology, (c) simulation methodology for transient breakthroughs in fixed beds, (d) data on pure component isotherms, (e) IAST calculations of mixture adsorption equilibria, and (f) data on transient breakthroughs in fixed bed adsorbers.
2. Separation of mixtures of 1-alcohols using CHA and SAPO-34
Let us examine the data on the pure component isotherms for a series of 1-alcohols in CHA, which is a cage type zeolite that consists of 316 Å3 sized cages separated by 3.8 Å × 4.2 Å sized windows. CBMC simulations of pure component 1-alcohols with C atoms in the 1–6 range in CHA at 300 K, as reported in earlier work,14 are shown in Fig. 2; the ESI† provides a brief summary of the CBMC methodology. The saturation capacity, Θi,sat, decreases from 5.4 molecules per cage for methanol to 1 molecule per cage for 1-hexanol.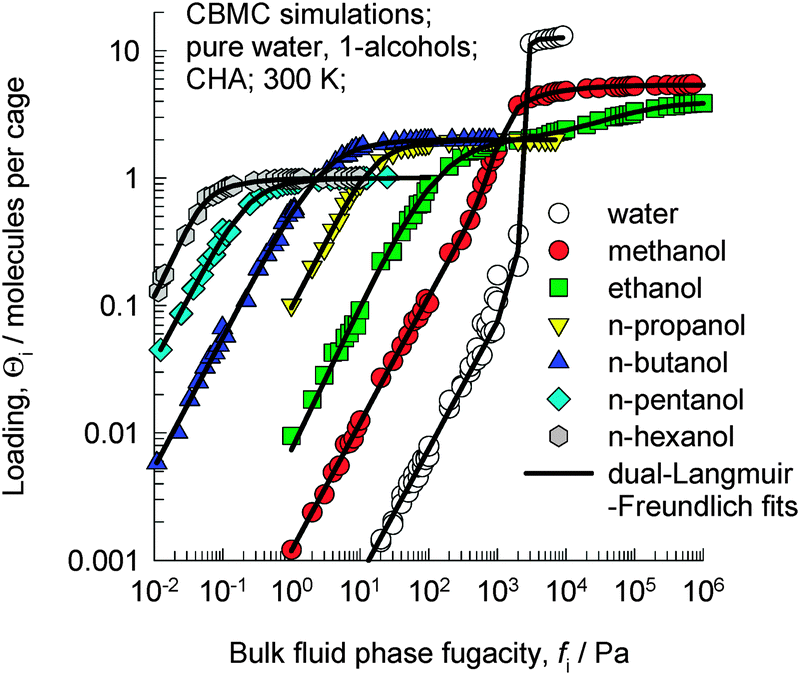 | ||
| Fig. 2 CBMC simulations14 of pure component adsorption isotherms for water and 1-alcohols in CHA at 300 K. | ||
The CBMC simulations for ethanol–1-propanol mixtures are shown in Fig. 3a. For total fluid phase fugacities ft < 300 kPa, the adsorption selectivity is strongly in favor of the longer 1-propanol molecule. However, when the total fluid phase fugacity ft exceeds 600 kPa, we find a reversal of selectivity. This selectivity reversal is entropy-based and is ascribable to the significantly higher saturation capacity of ethanol (4 molecules per cage) in comparison to that of 1-propanol (2 molecules per cage). The shaded region in Fig. 3a indicates that the bulk fluid phase is in the liquid phase for the range of fugacities fi. This region has been estimated using the Peng–Robinson equation of state. The CBMC data for mixture adsorption indicate that selectivity reversal is ensured when adsorption is from the liquid phase mixture and under these conditions the adsorption favors the alcohol with the shorter chain length.
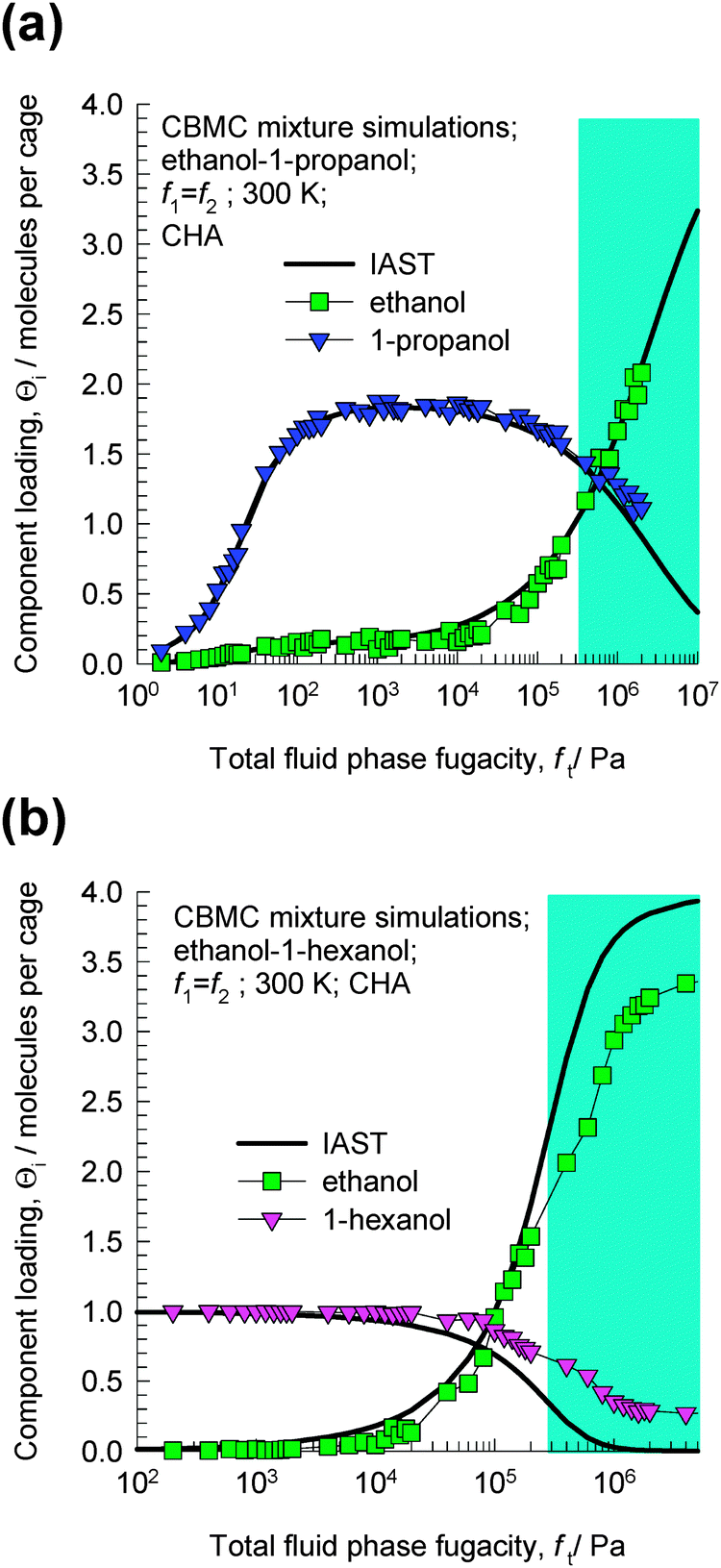 | ||
| Fig. 3 CBMC mixture simulations14 for (a) ethanol–1-propanol and (b) ethanol–1-hexanol in CHA at 300 K. The partial fugacities in the bulk fluid phase are taken to be equal, i.e. f1 = f2. The continuous solid lines represent calculations of the ideal adsorbed solution theory (IAST).15 | ||
The CBMC simulations for ethanol–1-hexanol mixtures are shown in Fig. 3b. For total fluid phase fugacities ft < 100 kPa, the adsorption selectivity is strongly in favor of the longer 1-hexanol molecule. However, when the total fluid phase fugacities ft exceed 200 kPa, we find a reversal of selectivity. This selectivity is entropy-based and is ascribable to the significantly higher saturation capacity of ethanol (4 molecules per cage) in comparison to that of 1-hexanol (1 molecule per cage). The shaded region in Fig. 3b indicates that the bulk fluid phase is in the liquid phase for the range of fugacities fi.
The continuous solid lines in Fig. 3a and b are the predictions of the ideal adsorbed solution theory (IAST) of Myers and Prausnitz15 using pure component isotherm fits. The IAST calculations have been presented here to demonstrate that selectivity reversal is not an unexpected phenomenon, but is a natural result that is obtained for a mixture of two species having (1) lower adsorption strength, but higher saturation capacity, and (2) higher adsorption strength, but lower saturation capacity. When saturation conditions are approached the component with the higher saturation capacity may be preferred. This is due to the fact that vacant “sites” are more easily filled by the smaller molecule under near-saturation conditions.
Remy et al.16 report experimental data on transient breakthroughs of (a) ethanol–1-propanol and (b) ethanol–1-hexanol mixtures in a fixed bed adsorber packed with SAPO-34, that has the same structural topology as CHA; see Fig. 4. The experiments carried out in the liquid phase are quite remarkable because the component that is eluted first from the adsorber is the alcohol with the longer chain length. The rationalization of these experimental data can be traced to the entropy effects that favor the shorter alcohols under conditions such that the bulk fluid phase is in the liquid state; cf.Fig. 3. When operating under conditions such that the bulk fluid phase is a liquid mixture, both adsorption and diffusion favor the uptake of the shorter alcohol, and the longer alcohol is rejected in a fixed bed adsorber. Both adsorption and intra-crystalline diffusion act synergistically in such separations; further explanations are provided in earlier reports.10,17 Since molecules jump one-at-a-time across the narrow windows of CHA and SAPO-34, slowing-down effects due to correlations are not likely to be significant.
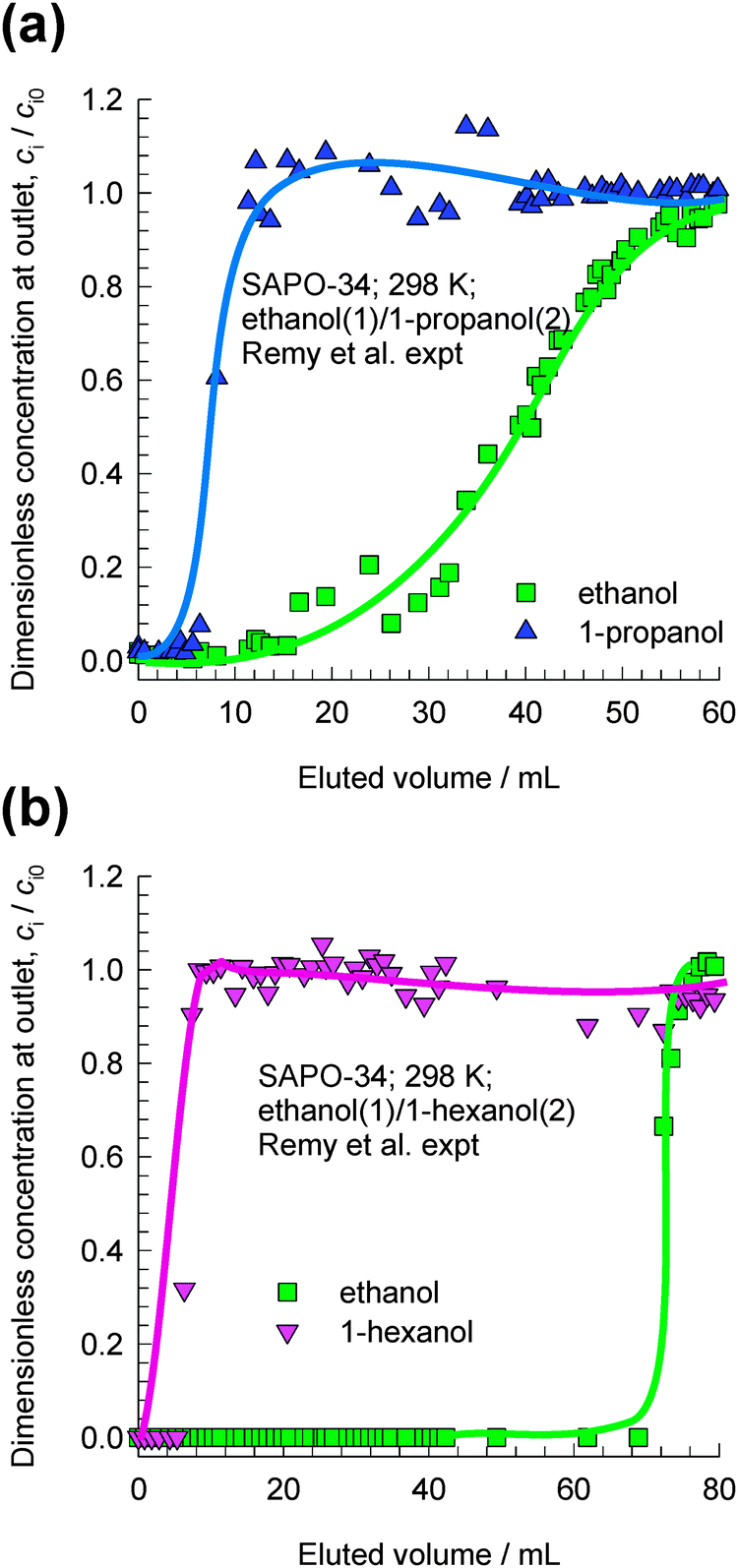 | ||
| Fig. 4 Transient breakthrough experimental data of Remy et al.16 for separation of (a) ethanol–1-propanol and (b) ethanol–1-hexanol mixtures in a fixed bed adsorber packed with SAPO-34. In their experiments the bulk mixtures were in the liquid state. The x-axis is the dimensionless time, as defined in the ESI.† | ||
The selective adsorption of the molecules with shorter chain lengths, in preference to the ones with longer chain lengths, has wider practical applications; the same principle can be applied to separate linear alkanes with chain lengths of up to 25 C atoms.14,18 For longer chain lengths, the chosen zeolite or MOF must have larger cages, such as TSC zeolite with a cage diameter of 17 Å.14
3. Separation of mixtures of 1-alcohols and water using ZIF-8
ZIF-8 has the SOD (sodalite) topology and consists of 1168 Å3 sized cages separated by 3.3 Å sized windows. In view of the larger cage capacity of ZIF-8 compared to CHA, we should expect the saturation loadings to be significantly higher. Fig. 5 shows the experimental data of Zhang et al.19 of pure component adsorption isotherms for 1-alcohols in hydrophobic ZIF-8 at 308 K. The saturation capacities are as follows: methanol ≈16 molecules per cage; ethanol ≈9 molecules per cage; 1-propanol ≈7 molecules per cage; 1-butanol ≈6 molecules per cage.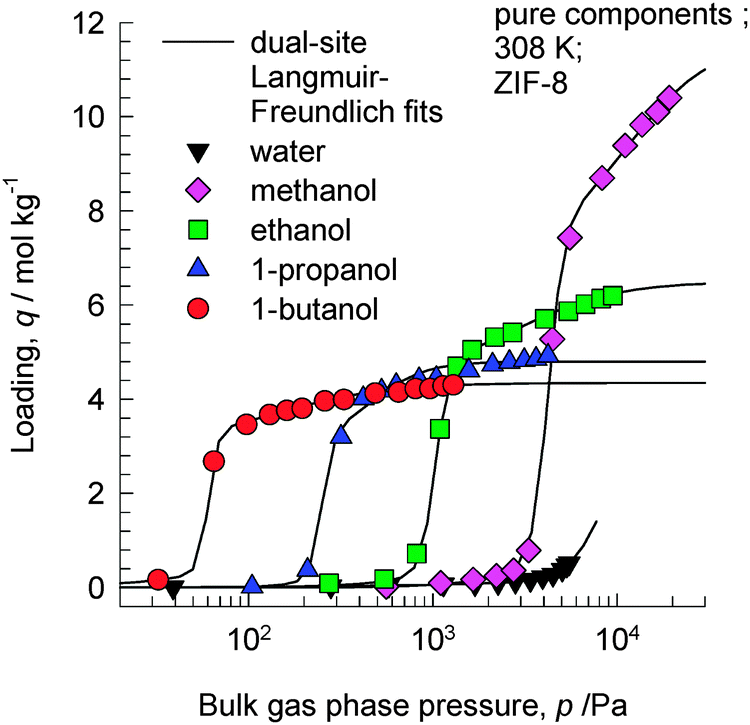 | ||
| Fig. 5 Experimental data of Zhang et al.19 for pure component adsorption isotherms for 1-alcohols in ZIF-8 at 308 K. Isotherm fit data are provided in the ESI.† | ||
IAST calculations of the component loading in ZIF-8 in equilibrium with a bulk fluid phase containing an equimolar methanol–1-ethanol–1-propanol–1-butanol mixture are shown in Fig. 6. At total pressures below 10 kPa, the hierarchy of component loadings is “normal”, i.e. decreases with decreasing chain length: 1-butanol > 1-propanol > ethanol > methanol. At pressures above 300 kPa, the hierarchy of component loadings is reversed, and increases with decreasing chain length: 1-butanol < 1-propanol < ethanol < methanol. The shaded region in Fig. 6 indicates that the bulk fluid phase is in the liquid phase. The IAST calculations show that for separations of liquid mixtures, the adsorption selectivity is in favor of the component with the shorter chain length, precisely analogous to the results for CHA zeolite presented in Fig. 3.
 | ||
| Fig. 6 IAST calculations of the component loading in ZIF-8 in equilibrium with a bulk fluid phase containing an equimolar methanol–1-ethanol–1-propanol–1-butanol mixture at 308 K. The range of liquid phase operation is indicated by the shaded region; the transition between vapor and liquid bulk phase is determined using the Peng–Robinson equation of state. | ||
Due to the hydrophobicity of ZIF-8, the adsorption loading of water is significantly lower than that of 1-alcohols; see Fig. 5. Consequently, ZIF-8 has significant potential for use in recovery of alcohols from biofuel products that, typically, are dilute alcohol-in-water solutions. In such recovery processes, it is essential to use materials that selectively adsorb the alcohols and reject water. Fig. 7 shows transient breakthrough simulations for the 1-alcohols–water mixture using ZIF-8. The inlet consists of 90% water, containing 2.5% each of methanol, 1-ethanol, 1-propanol, and 1-butanol at a total pressure of 105 Pa. As desired, water is the first component to be rejected. Subsequently, the sequence of breakthroughs is methanol, ethanol, 1-propanol, and 1-butanol that corresponds to the adsorption strengths in the Henry regime. It must be emphasized that the separation of dilute alcohol–water mixtures is based on adsorption selectivities in the Henry regime and not on molecular packing effects. Molecular packing effects are in play only near pore saturation conditions.
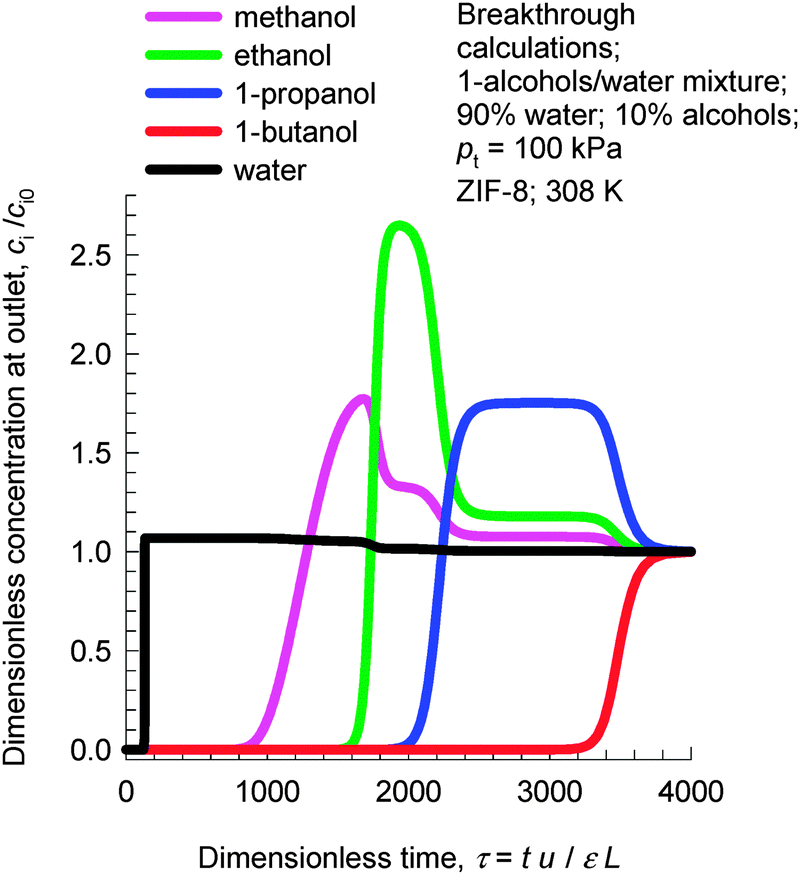 | ||
| Fig. 7 Transient breakthrough simulations for 1-alcohols–water mixture using ZIF-8. The inlet feed consists of a 25/25/25/25/900 methanol/1-ethanol/1-propanol/1-butanol/water mixture at 308 K at a total pressure of 100 kPa. | ||
The separation performance of ZIF-8 for water–alcohol separations is expected to be significantly superior to that of CHA or SAPO-34 because of its significantly larger cage volume. Consequently, the saturation loadings are higher in magnitude; this leads to longer breakthrough times in fixed bed adsorption devices.10
4. Separation of mixtures of 1-alcohols using ZIF-68
Van der Perre et al.20 have investigated separations with ZIF-68 whose structure consists of ZnN4 tetrahedra that are connected by both polar 2-nitroimidazole (nIM) ligands and nonpolar benzimidazole (bIM) ligands. It has the same topology as GME zeolite, with 1D channels of two different sizes; see ESI† for pore landscapes. Fig. 8a shows the experimental data of Van der Perre et al.20 for pure component adsorption isotherms of 1-alcohols in ZIF-68 at 323 K. With increasing chain length, the saturation capacities are significantly lowered. | ||
| Fig. 8 (a) Experimental data of Van der Perre et al.20 for pure component adsorption isotherms of 1-alcohols in ZIF-68 at 323 K. (b) IAST calculations of the component loadings for an equimolar 6-component methanol–ethanol–1-propanol–1-butanol–1-pentanol–1-hexanol mixture. | ||
Fig. 8b shows IAST calculations of the component loadings for an equimolar 6-component methanol–ethanol–1-propanol–1-butanol–1-pentanol–1-hexanol mixture. At pressures below about 1 MPa, the adsorption loadings increase with increasing chain length. However, for pressures above about 2 MPa the component loading decreases with increasing chain length; this is because of entropy effects that favor shorter 1-alcohols. At 323 K, the bulk fluid phase is in the liquid state for pt > 1 MPa; this implies that for adsorption of liquid phase mixtures, entropy effects will favor the shorter alcohol.
As an example, let us consider transient uptake in ZIF-68 crystals of a binary equimolar ethanol–1-butanol mixture at 323 K. Fig. 9a and b show simulations of transient uptake within ZIF-68 crystals exposed to a bulk gas phase pressure of (a) 400 kPa and (b) 4 MPa. The values of the intra-crystalline diffusivities used in the simulations are based on the experimental data of Van der Perre et al;20 the diffusivity of ethanol is 25 times higher than that of 1-butanol. For bulk fluid pressure pt = 400 kPa, the uptake of 1-butanol at equilibrium is about twice that of ethanol. However, for fluid pressure pt = 4 MPa, the uptake of 1-butanol at equilibrium is 60% that of ethanol; Fig. 9b. Van der Perre et al.20 have experimentally measured the uptake of ethanol–1-butanol liquid phase mixtures in ZIF-68 and found results that are in qualitative agreement with those presented in Fig. 9b. The experimental uptake data of Van der Perre et al.20 offer convincing evidence of the entropy effects that influence separations of mixtures of 1-alcohols in ZIF-68.
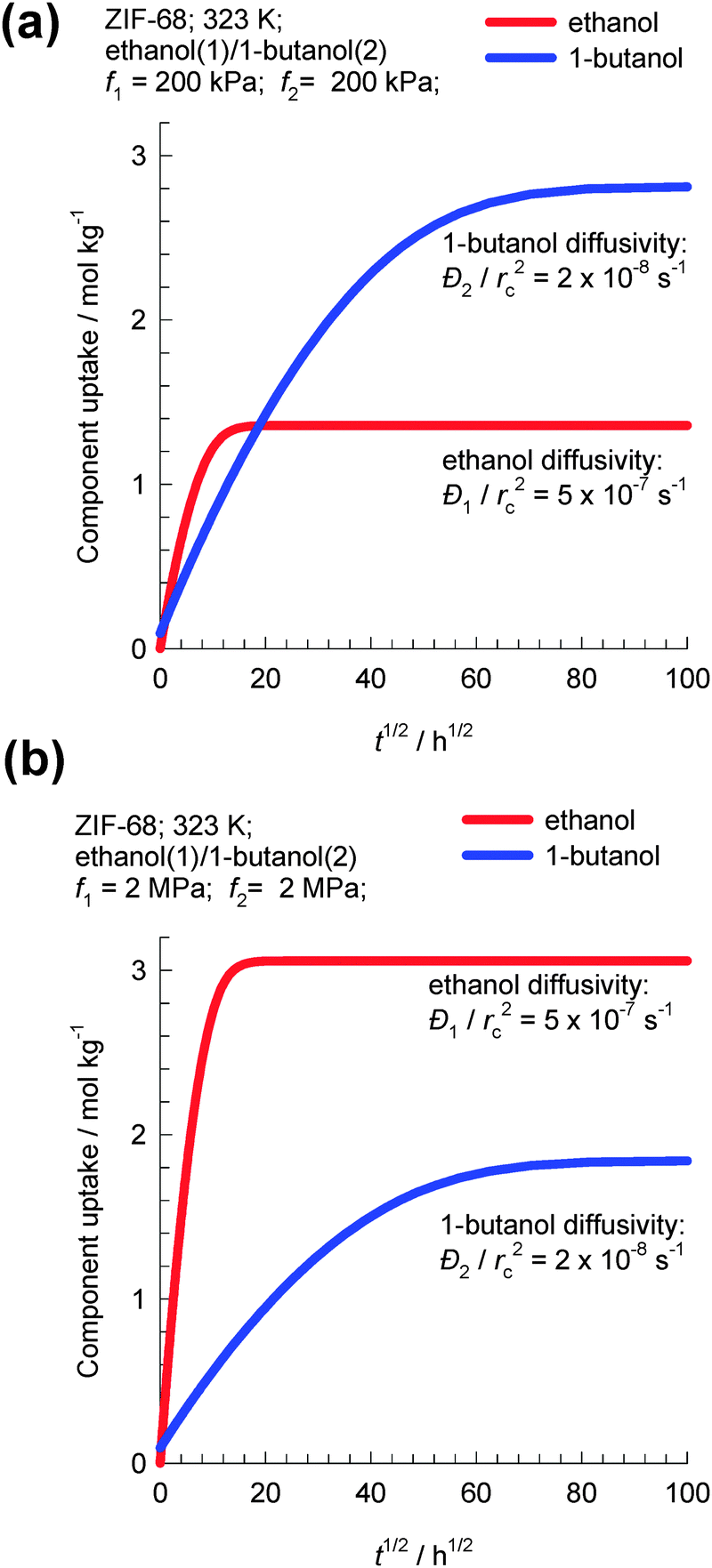 | ||
| Fig. 9 Transient uptake within ZIF-68 crystals exposed to a bulk gas phase pressure of (a) 400 kPa and (b) 4 MPa. Simulation details are provided in the ESI.† | ||
A further important aspect of the data in Fig. 9b is that both adsorption and diffusion favor the shorter 1-alcohol, i.e. there is synergy between adsorption and diffusion. This synergy arises because of the combination of two reasons: (1) the adsorption of the shorter alcohol is preferred because of molecular packing effects and (2) the diffusivity of the shorter alcohol is higher based on the diffusivities reported by Van der Perre et al.20 It must however be remarked that the diffusivity data of Van der Perre et al.20 were measured at low occupancies, and these data would be relevant at pore saturation provided slowing-down effects are not significant.10 Generally speaking, slowing-down effects will work against the synergistic aspects.
5. Separation of water–benzene mixtures using CuBTC
The structure of CuBTC (Cu3(BTC)2, also known as HKUST-1) consists of two types of “cages” and two types of “windows” separating these cages. The large cages are inter-connected by 9 Å windows of square cross-section. The large cages are also connected to tetrahedral-shaped pockets of ca. 6 Å size through triangular-shaped windows of ca. 4.6 Å size. The tetrahedral pockets can accommodate small molecules such as water but larger molecules can be excluded in these pockets; they must locate only in larger cages.21
Let us consider the separation of equimolar water(1)–benzene(2) vapor phase mixtures using CuBTC. Fig. 10a presents IAST calculations of the adsorption selectivities as a function of the total bulk pressure pt using the pure component isotherm data from Zhao et al.22 For operations at T > 308 K and pressures pt < 10 kPa, the selectivity is in favor of benzene which has a higher adsorption strength, resulting from π electron exchanges between the benzene rings of the guest molecules and the unsaturated Cu atoms of CuBTC. High temperatures and low pressures imply low pore occupancies. The situation is dramatically different when we operate at pressures pt > 10 kPa; the selectivity is in favor of water, which has a higher saturation capacity.
 | ||
| Fig. 10 IAST calculations of water–benzene adsorption selectivities for equimolar water–benzene mixtures in CuBTC at temperatures of 288 K, 298 K, 308 K, and 318 K. Further calculation details are provided in the ESI.† | ||
In Fig. 10b the adsorption selectivity is plotted as a function of the total mixture loading, qt = q1 + q2. We note from the data in Fig. 10b that at mixture loadings qt > 18 mol kg−1, the influence of temperature is practically negligible. Furthermore, the data show that for qt > 18 mol kg−1, the selectivity is in favor of water by about two orders of magnitude. The selectivities are in favor of water because of entropy effects, which are governed by saturation capacities.
Zhao et al.22 have presented transient breakthrough experimental data that confirm the expectation of water-selective adsorption for conditions such that molecular packing effects are dominant. It must however be remarked that Zhao et al.22 do not explain their results in terms of entropy effects as we have done.
Though no experimental data are reported as yet, we should expect CuBTC also to be effective for separation of water–alcohol mixtures. An important advantage of CuBTC in such separations is its larger pore volume (0.86 cm3 g−1), which is higher than that of ZIF-8 (0.5 cm3 g−1), ZIF-68 (0.439 cm3 g−1), and ZIF-71 (0.736 cm3 g−1). Larger pore volume results in higher saturation capacities and better separations in fixed bed adsorbers because the breakthrough times will be longer.
6. Separation of mixtures containing water, methanol, and ethanol using ZIF-71, FAU, DDR, MFI, FER, and LTA-4A
ZIF-71 possesses a three-dimensional pore network formed by large cages interconnected via small windows.23 The CBMC simulations of Nalaparaju et al.23 for water–methanol and water–ethanol mixtures in ZIF-71 provide convincing evidence of the entropy effects that manifest under pore saturation conditions. CBMC simulations of the pure component isotherms of water, methanol and ethanol in ZIF-71 at 298 K are shown in Fig. 11a. The significantly higher saturation capacity of water compared to that of methanol and ethanol is evident. Fig. 11b and c shows CBMC simulations of the component loadings for equimolar (b) water–methanol and (c) water–ethanol mixtures in ZIF-71. For total pressures below 20 kPa, the adsorption is in favor of the alcohol. However, as the total pressure increases above 20 kPa, entropy effects cause the water loadings to exceed that of the partner alcohol molecules. At 100 kPa, the water–methanol and water–ethanol adsorption selectivities approach values of about 5. These results imply that for adsorption from liquid phase mixtures, water can be selectively adsorbed from water–alcohol mixtures.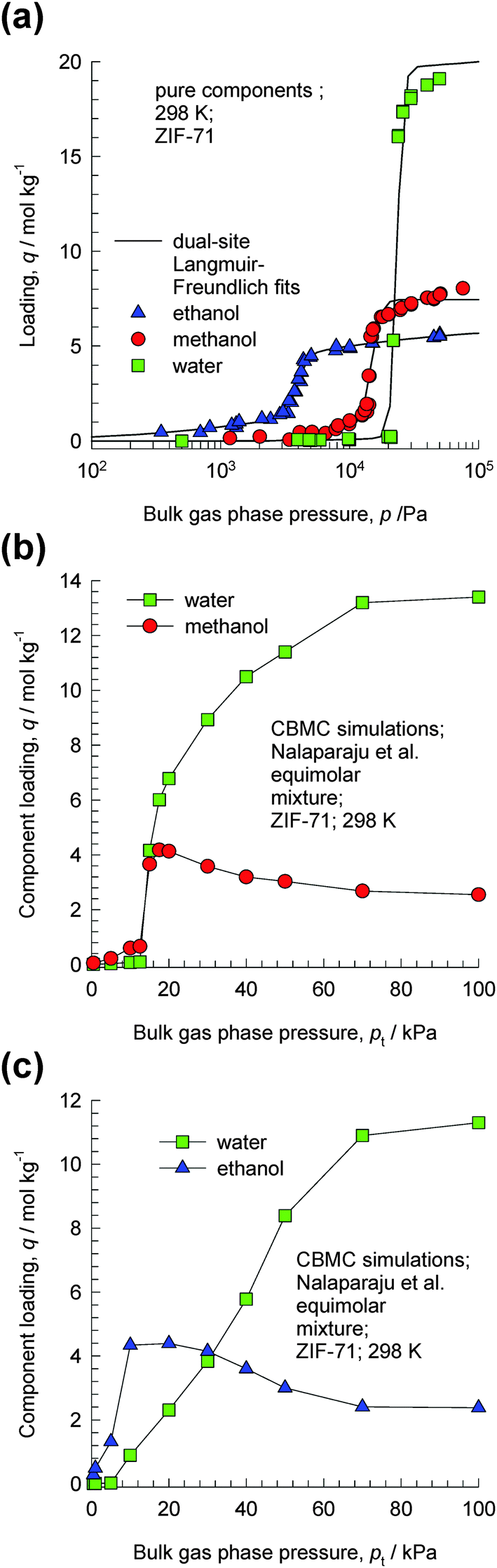 | ||
| Fig. 11 (a) CBMC simulation data of Nalaparaju et al.23 for pure component adsorption isotherms of water (only the “adsorption” branch of the isotherms is shown here), methanol and ethanol in ZIF-71 at 298 K. (b, c) CBMC simulations of the component loadings for equimolar (b) water–methanol and (c) water–ethanol mixtures in ZIF-71. | ||
Further evidence of selectivity reversals at pore saturation for adsorption of water–methanol and water–ethanol mixtures is available in the published literature24 for FAU, DDR, MFI, LTA-4A, and FER which are hydrophilic zeolites; the information is summarized in the ESI.† These data demonstrate that IAST predicts such selectivity reversals reasonably accurately. The ESI† also provides pore landscapes and snapshots that will assist the reader in appreciating how molecules pack within the pores. For water-selective adsorption, MD simulations of self-diffusivities indicate that intra-crystalline diffusion effects serve to enhance the separations in favor of water for hydrophilic zeolites.
Let us consider the LTA zeolite for which the all-silica form (i.e. without cations, also called ZK-4) consists of 743 Å3 cages that are separated by windows of approximately 4.1 Å × 4.7 Å. In LTA-4A (96 Si, 96 Al, 96 Na+, Si/Al = 1), some of the Na+ cations partially block the window regions,25–29 thereby effectively reducing the aperture size that is available for inter-cage hopping of molecules. The Na+ and Ca++ cations in LTA-5A (96 Si, 96 Al, 32 Na+, 32 Ca++, Si/Al = 1), on the other hand, do not locate near the window regions and there is no blocking. This implies that diffusional influences are much stronger in LTA-4A than in LTA-5A zeolite. The presence of bulkier K+ cations in LTA-3A (96 Si, 96 Al, 96 K+, Si/Al = 1) causes the window blocking effect to be significantly enhanced, when compared to LTA-4A. For this reason, LTA-3A zeolite is used for selective removal of water from gaseous streams (dehumidification) and water–alcohol mixtures (dehydration).30 It has to be mentioned that it is common to find information in the published literature that suggests that 3A, 4A, and 5A zeolites have window apertures, respectively, of 3 Å, 4 Å, and 5 Å; this is not precisely correct. The degree of blocking (by the cations Na+ or K+) of the window apertures of the pristine framework with 4.1 Å × 4.7 Å decreases in the order as we progress from 3A, 4A to 5A.
An important application of LTA-4A is separation of water–ethanol mixtures by pervaporation. Conventional distillation of ethanol–water mixtures can produce ethanol with a purity close to 95 wt% owing to azeotrope formation. For obtaining say 99.5% pure ethanol, we need to feed the obtained 95 wt% ethanol product to an azeotropic distillation column with an entrainer such as benzene or cyclohexane. A better alternative, avoiding the use of entrainers, is to adopt a hybrid scheme (see Fig. 12) in which the 95 wt% ethanol top product is fed to a hydrophilic LTA-4A zeolite membrane pervaporation unit. The desired 99.5% pure ethanol product is recovered as retentate. Water has a significantly higher diffusivity than ethanol due to the narrow window aperture; both adsorption and diffusion in LTA-4A favor permeation of water across the membrane.
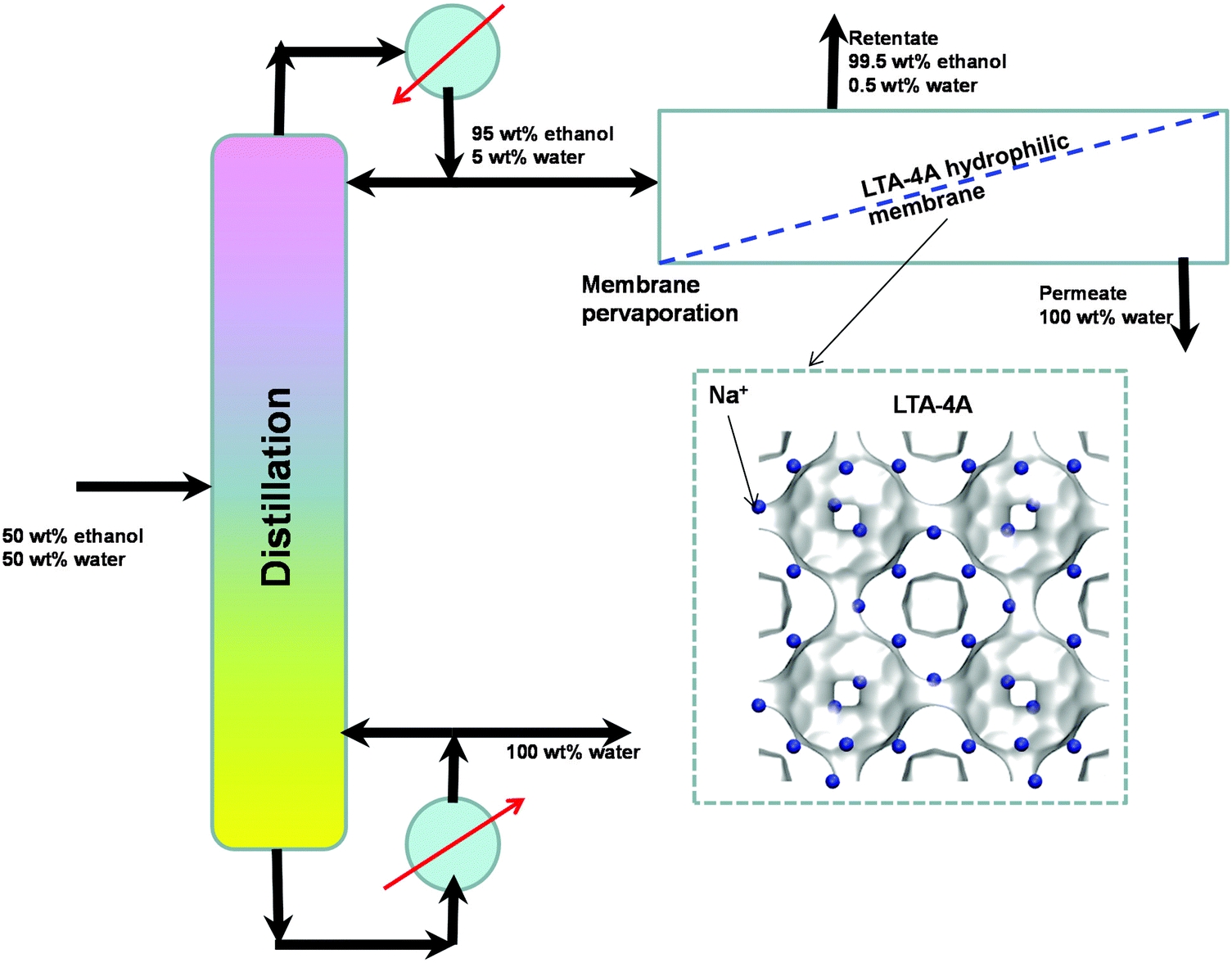 | ||
| Fig. 12 Selective water permeation across the LTA-4A zeolite membrane used in the hybrid scheme for production of 99.5% pure ethanol. | ||
There is considerable scope for synthesizing hydrophilic MOFs that have separation characteristics that are superior to those of LTA-4A. The pore dimensions and pore volumes need to be carefully chosen so as to offer larger saturation capacities, while retaining the synergy between adsorption and diffusion; this is a fruitful area for further research.
7. Separation of methanol–ethanol mixtures with UCY-5 and Ce(BTB)
Efthymiou et al.31 report experimental data on the uptake of methanol–ethanol mixtures in the liquid phase using a flexible lanthanide MOF, UCY-5, with Ce3+ as the metal ions. Transient uptake data of the components from binary 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 methanol–ethanol liquid phase mixtures are plotted in Fig. 13a and b. At long times, i.e. as equilibrium is approached, we note that the component loadings of methanol are significantly higher than those of ethanol, even in the case of 1:2 bulk liquid phase mixtures. This provides conclusive proof that the adsorption is strongly in favor of methanol which has a shorter chain length. We believe that their experiments are a clear confirmation of the molecular packing effects that favor methanol over ethanol. Furthermore, we note that the uptake of ethanol is significantly slower than that of methanol, by about an order of magnitude. This implies that both adsorption and diffusion are synergistically in favor of methanol. This synergistic effect is, in principle, analogous to that observed for mixtures of 1-alcohols in SAPO-34.16
:1 and 1:2 methanol–ethanol liquid phase mixtures are plotted in Fig. 13a and b. At long times, i.e. as equilibrium is approached, we note that the component loadings of methanol are significantly higher than those of ethanol, even in the case of 1:2 bulk liquid phase mixtures. This provides conclusive proof that the adsorption is strongly in favor of methanol which has a shorter chain length. We believe that their experiments are a clear confirmation of the molecular packing effects that favor methanol over ethanol. Furthermore, we note that the uptake of ethanol is significantly slower than that of methanol, by about an order of magnitude. This implies that both adsorption and diffusion are synergistically in favor of methanol. This synergistic effect is, in principle, analogous to that observed for mixtures of 1-alcohols in SAPO-34.16
 | ||
| Fig. 13 Experimental data of Efthymiou et al.31 for transient uptake of (a) 1:1 methanol–ethanol and (b) 1:2 methanol–ethanol mixtures in UCY-5. The data in Fig. 9a and b of Efthymiou et al.31 are re-plotted here using the square root of time (in minutes) on the x-axis. (c) IAST calculations of component loadings for equimolar methanol–ethanol mixtures in Ce(BTB) at 298 K. (d) Simulations of transient uptake of equimolar methanol–ethanol mixtures in Ce(BTB) at a total pressure of 100 kPa. Further simulation details are available in the ESI.† | ||
Lin et al.32 present experimental isotherm data for methanol, ethanol, 1-propanol, 2-propanol, and 1-butanol in a homochiral ultramicroporous lanthanide–organic framework, Ce(BTB) (H3BTB = 1,3,5-benzenetrisbenzoic acid). Their data show the following hierarchy of saturation capacities: methanol ≫ ethanol > 1-propanol > 1-butanol ≫ 2-propanol. This hierarchy of saturation capacities implies that it is possible to separate mixtures of 1-alcohols based on the chain length. As illustration, Fig. 13c shows IAST calculations of component loadings for equimolar methanol–ethanol mixtures in Ce(BTB) at 298 K. For total pressures greater than 20 kPa, the selectivity is in favor of methanol because of its higher saturation capacity. Simulations of transient uptake of methanol–ethanol mixtures within the crystals of Ce(BTB) at a total pressure of 100 kPa are shown in Fig. 13d, from which it is clear that methanol-selective characteristics of Ce(BTB) are similar to those observed for UCY-5.
Breakthrough experiments need to be undertaken in order to investigate the separation performance of Ce(BTB) in more detail.
8. Separation of mixtures of water and alcohols with Co(pbdc)
Fig. 14a presents the pure component isotherms for water, methanol, ethanol, 1-propanol, and 2-propanol at 298 K reported by Zheng et al.33 in Co(pbdc) with a 2-fold interpenetrating net. For pressures greater than 10 kPa, the hierarchy of component loadings is water > methanol > ethanol > 1-propanol > 2-propanol. For adsorption from water–alcohol liquid phase mixtures, we should expect the selectivity to be in favor of water. Fig. 14b shows the transient breakthrough simulations for a water–ethanol mixture with an azeotropic composition of 11 mol% water. The breakthrough simulations indicate that the azeotropic mixture can be separated in a fixed bed adsorber packed with Co(pbdc). Similarly, the transient breakthrough simulations in Fig. 14c and d demonstrate that water–1-propanol and water–2-propanol mixtures of azeotropic compositions can be separated with Co(pbdc). | ||
| Fig. 14 (a) Pure component isotherms for water, methanol, ethanol, 1-propanol, and 2-propanol at 298 K in Co(pbdc).33 (b–d) Transient breakthrough simulations for (b) 11/89 water/ethanol, (c) 57/43 water/1-propanol, and (d) 31/69 water/2-propanol mixtures in a fixed bed adsorber packed with Co(pbdc) at 298 K and 100 kPa. (e, f) Transient breakthrough simulations for 20/80 water/methanol and (b) 11/89 water/ethanol mixtures in a fixed bed adsorber packed with CID-1 at 298 K and 100 kPa. Video animations showing the transient breakthrough of water–ethanol mixtures in beds packed with CID-1 and Co(pbdc) are available as ESI.† | ||
Analogous water-selective separations of water–alcohol mixtures are possible with CID-1,34 Cu-GLA,35 Zn(II)-MOF,36,37 and JUC-110;38 detailed analyses are provided in the ESI.†
It must be stressed that we have examined the separations of water–alcohol mixtures with Co(pbdc), CID-1, Cu-GLA, Zn(II)-MOF, and JUC-110 solely on the basis of pure adsorption isotherms. Gu et al.37 have reported the selective uptake of water from various organic compounds (ethanol, acetone, tetrahydrofuran, benzene, toluene, xylene) using Zn(II) and a rigid planar ligand IDC3 (imidazole-4,5-dicarboxylate); this MOF consists of a 3D porous metal–organic framework (MOF) with 1D open channels. The selective adsorption of water reported with this Zn(II) MOF is most likely due to entropy effects, but the authors themselves do not offer such an explanation.
The breakthrough characteristics, presented in the ESI,† appear to indicate that the porous interdigitated coordination polymer CID-1 is superior to other MOFs, mainly due to its higher water uptake capacity. To demonstrate this, Fig. 14e and f show the transient breakthroughs of water–methanol and water–ethanol mixtures in fixed beds packed with CID-1. Comparison of Fig. 14b and f shows that the breakthrough of water occurs significantly later with CID-1 than with Co(pbdc); this is desirable. Also, we note that ethanol breaks through earlier with CID-1 than with Co(pbdc); this indicates much better separations using CID-1. There is a need to perform breakthrough experiments to confirm the potential of CID-1. We also need to search for or synthesise MOFs with characteristics that are superior to those of CID-1. In view of the importance of water–alcohol separations in industry, this research has the potential of offering energy reduction opportunities.
9. Separation of C8 aromatics
Aromatic hydrocarbons, that are valuable feedstocks in the petrochemical industries, are most commonly obtained from catalytic reforming of naphtha. The xylene isomers, o-xylene, m-xylene and in particular p-xylene, are important chemical intermediates. ortho-Xylene is oxidized to make phthalic anhydride which is used to make phthalate plasticizers among other things. meta-Xylene is oxidized to produce isophthalic acid, which is used in unsaturated polyester resin. However, p-xylene has the largest market of the three isomers; the demand for p-xylene is several times that of m-xylene and o-xylene. The largest use of p-xylene is in its oxidation to make terephthalic acid, which is used in turn to make polymers such as polyethylene terephthalate (PET) and polybutylene terephthalate (PBT). PET is one of the largest volume polymers in the world, and is used to produce fibers, resins, films, and blown beverage bottles.In a commonly used separation scheme (cf.Fig. 15), the xylene rich stream from the bottom of the reformer splitter is routed to a xylene splitter. Here, the heavier aromatics (C9+) are removed from the bottom of the column. The overhead stream from the xylene splitter containing o-xylene, m-xylene, p-xylene, and ethylbenzene needs to be separated for recovery of p-xylene. Due to the very small differences in boiling points, p-xylene recovery from o-xylene–m-xylene–p-xylene–ethylbenzene mixtures is not possible by use of distillation technology. There are, however, significant differences in the freezing points (see Fig. 16) that allow fractional crystallization to be used for separations. The differences in the freezing points arise because of differences in the stacking efficiencies of molecules. para-Xylene has the highest freezing point because these molecules stack most efficiently; pure p-xylene crystals are the first to emerge from the solution upon cooling. However, the energy requirements for fractional crystallization are high because of the need to cool to temperatures of about 220 K. Selective adsorption of xylene isomers within the pores of ordered crystalline microporous materials is an energy-efficient alternative to fractional crystallization. In currently used technology the separation is carried out using cation-exchanged Faujasite (FAU) zeolite in a simulated moving bed (SMB) adsorption separation unit. An SMB unit consists of a set of interconnected columns in series; countercurrent flow of the solid and liquid phases is simulated by the periodic shifting of the inlets and outlets in the direction of the liquid flow. Commonly used SMB technologies are UOP's Parex, Axens' Eluxyl, and Toray's Aromax.12,39 The typical composition of a mixed xylene feed to a simulated moving bed (SMB) adsorber is 19% ethylbenzene, 44% m-xylene, 20% o-xylene, and 17% p-xylene. Since the adsorbent particles are in contact with a mixture in the liquid phase, the pores of the adsorbent material are practically saturated with guest molecules (cf.Fig. 1d). The hierarchy of adsorption strengths is dictated by molecular packing or entropy effects. Binding energies of guest molecules with the framework walls or non-framework cations do not solely determine the separation performance. As pointed out by Peralta et al.,40 adsorbents selective to p-xylene are desirable for high productivities; they need to adsorb only ∼20% of the feed, whereas an adsorbent that rejects p-xylene would have to adsorb 80% of the feed. In current industrial practice the adsorbent used is BaX zeolite, which selectively adsorbs p-xylene. Typically, BaX zeolite also contains other cations such as K+. The separation of xylenes at pore saturation in BaX is influenced also by factors other than molecular packing effects.
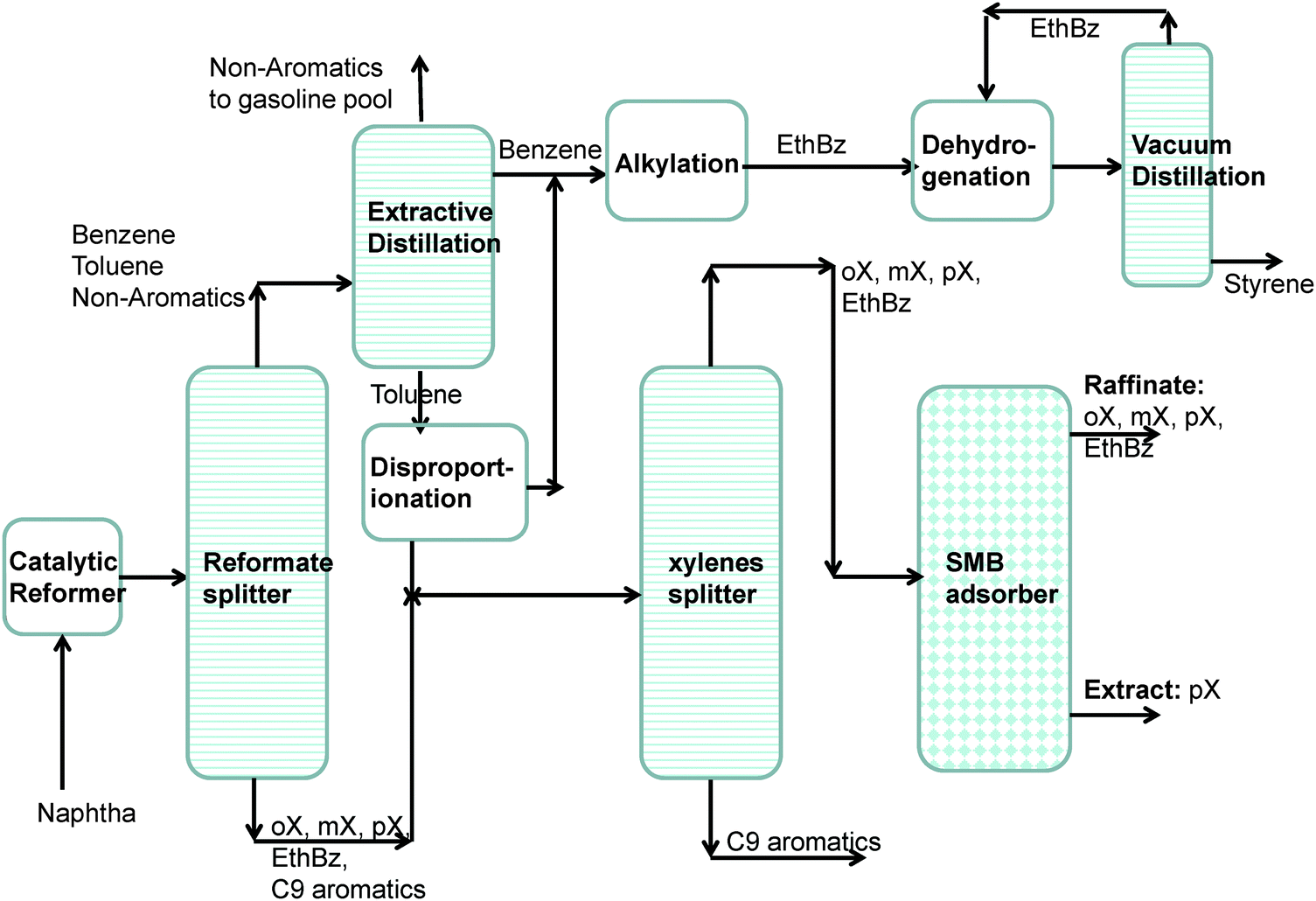 | ||
| Fig. 15 Schematic showing the separations of the products from a catalytic reforming unit. | ||
 | ||
| Fig. 16 Boiling points and freezing points of C8 hydrocarbons, along with molecular dimensions. | ||
On examination of the boiling points of the C8 aromatics, we note that o-xylene has the highest boiling point (cf.Fig. 16). A xylene splitter, which ordinarily would separate the C9+ aromatics from the xylenes (cf.Fig. 15), can be redesigned to remove o-xylene as well; this alternative configuration is depicted in Fig. 17. In this configuration, the fractionation split in the xylene splitter is between m-xylene and o-xylene with a temperature difference of only 5 K; consequently we need a super-fractionating tower containing about 135 fractionating trays. The overhead product from the xylene splitter, rich in ethylbenzene, p-xylene, and m-xylene, is fed to a recovery unit for p-xylene. The bottom product from the super-fractionator is routed to a further distillation column in which o-xylene is separated from C9+ aromatics and recovered as an overhead product. In the configuration shown in Fig. 17, both the xylene splitter and o-xylene recovery columns are super-fractionators that have high energy demands.
 | ||
| Fig. 17 An alternative separation scheme in which o-xylene is recovered at the bottom of the reformate splitter. | ||
There is great potential for the discovery of MOFs that have higher selectivity to adsorb p-xylene than obtainable with BaX zeolite, used in the SMB adsorber in the separation schemes in Fig. 16 and 17. Since the process operates under pore saturation conditions, we need to find MOFs that have the highest saturation capacity for p-xylene. Therefore, we seek MOFs that have the proper channel dimensions to allow optimum packing of p-xylene molecules.
Both the distillation columns in the processing scheme shown in Fig. 17 could be replaced by an adsorption unit, packed with a MOF that selectively adsorbs o-xylene from aromatic mixtures.
To arrive at the optimum channel dimensions for selective adsorption of either p-xylene or o-xylene, let us first examine the molecule dimensions. The height and width of the C8 aromatics are as follows: o-xylene: 8 Å × 7.4 Å; m-xylene: 8.9 Å × 7.4 Å; p-xylene: 9.3 Å × 6.7 Å; ethylbenzene: 9.5 Å × 6.7 Å; styrene: 9.3 Å × 6.7 Å; see dimensions provided in Fig. 16. A further point to note is that xylene isomers are flat molecules; these isomers can align themselves parallel to the channel walls, affording better van der Waals interactions with the framework atoms. By contrast, ethylbenzene is not a flat molecule; the ethyl branch is not in the same plane as the benzene ring.
Due to the differences in the molecular dimensions of the xylene isomers, the efficiencies with which the xylene isomers stack within the channels of different dimensions are different. Stacking xylenes within 1D channels of MOFs is analogous to stacking books within bookshelves.10,41 Consider the stacking of o-xylene and p-xylene within one-dimensional (1D) channels that are 8.5 Å wide; see Fig. 18a and b. Within 8.5 Å channels, the stacking efficiency of o-xylene is higher than that of p-xylene, i.e. more o-xylene molecules can be packed within a channel of a given length. The channel size is not wide-enough to allow p-xylene to stack vertically, and fewer molecules of p-xylene can be packed within the same channel length.
 | ||
| Fig. 18 Cartoons showing stacking of (a) o-xylene and (b) p-xylene within the 1D 8.5 Å channels. | ||
Experimental data42–44 for MIL-47 and MIL-53 with 1D rhombohedric channels of 8.5 Å confirm that these MOFs are selective to adsorption of o-xylene when operating under conditions close to pore saturation. The snapshots in Fig. 19, obtained from CBMC simulations,41 clearly show the optimal stacking of o-xylene within 8.5 Å channels of MIL-47.
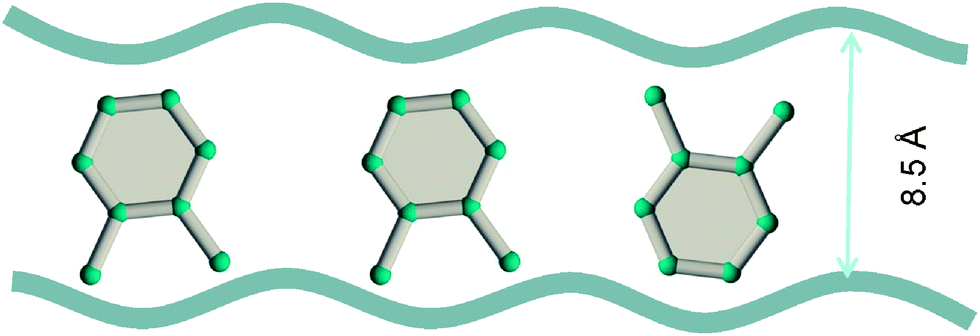 | ||
| Fig. 19 Snapshots, obtained from CBMC simulations,41 showing the stacking of o-xylene within 8.5 Å channels of MIL-47. | ||
Experimental data of Niekiel et al.45 for adsorption isotherms for xylene isomers in CAU-13 show strong selectivity towards o-xylene that has optimal stacking within the 8.46 Å channels. Fang et al.46 report pulse breakthrough simulations for 4-component o-xylene–m-xylene–p-xylene–ethylbenzene in MOF-CJ3 that indicate adsorption selectivity towards o-xylene. MOF-CJ3 has square channels of approximately 8 Å size which is perhaps sufficient for commensurate stacking of o-xylene.
The MOFs MIL-47, MIL-53, CAU-13, and MOF-CJ3 are potentially usable as partial replacement of the distillation columns in Fig. 17. Within the channels of these MOFs, the o-xylene molecules stack themselves parallel to the surrounding walls, along the 1D channel axis. It must be remarked, however, that their use in industry would require considerably more testing for stability and other aspects.
The work of Chiang et al.47 indicates that the aluminophosphate molecular sieve AlPO4-5 (with AFI zeolite topology) has the capability of separating xylene isomers; as an indication, Fig. 20 shows the experimental data for pure component isotherms for xylenes in AlPO4-5 at 303 K. The hierarchy of saturation capacities is o-xylene ≫ p-xylene ≈ m-xylene. On the basis of molecular dimensions of the isomers, Chiang et al.47 have argued that the differences in the saturation capacities reflect differences in packing arrangements within the 1D channels of AlPO4-5. For m-xylene and p-xylene either the “height” or “width” is too large to allow vertical alignment; the orientation of these isomers occurs at an inclination. The “height” and “width” of o-xylene are both small enough for this isomer to be stacked vertically, face-to-face, within the 1D channels. The orientations of ortho, meta, and para isomers of xylene are pictured in Fig. 4 of the paper by Chiang et al;47 see discussions on these cartoons shown in their paper. An important consequence of the differences in the orientation of isomers is that the “footprints” of o-xylene molecules are significantly shorter than the footprints of m-xylene and p-xylene molecules. Shorter footprints result in higher saturation capacities, as evidenced in the data in Fig. 20. However, it must be remarked that the arguments of Chiang et al.47 are not backed by snapshots from molecular simulations.
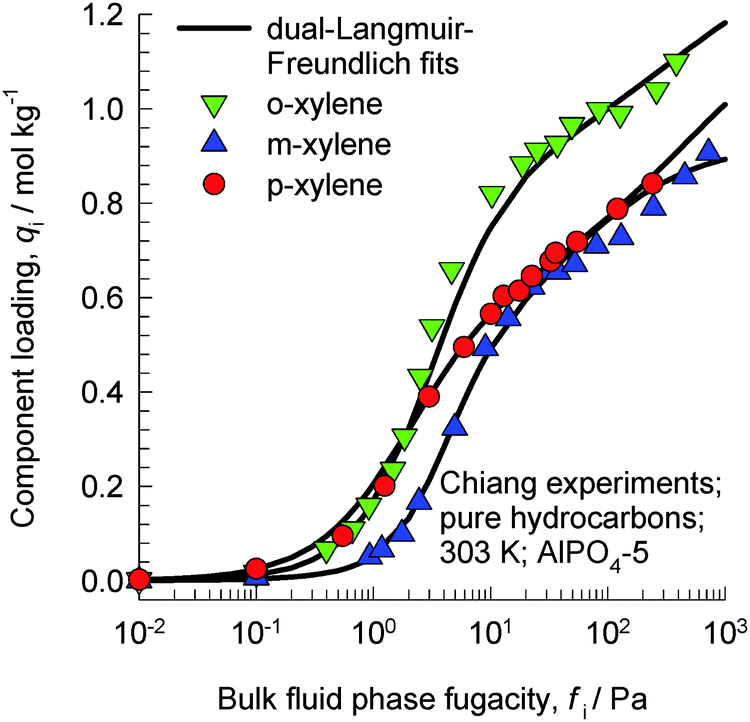 | ||
| Fig. 20 Experimental data of Chiang et al.47 for pure component isotherms for xylene isomers in AlPO4-5 at 303 K. | ||
IAST calculations of the adsorption equilibrium for 3-component o-xylene(1)–m-xylene(2)–p-xylene(3) mixtures at 303 K are shown in Fig. 21. We note that AlPO4-5 is p-xylene selective in the Henry regime, but becomes strongly o-xylene selective as pore saturation is approached.
 | ||
| Fig. 21 IAST calculations of adsorption selectivity in a 3-component o-xylene–m-xylene–p-xylene mixture in AlPO4-5 at 303 K. Calculation details are provided in the ESI.† | ||
Transient breakthroughs at a total xylene pressure pt = 30 kPa are shown in Fig. 22a; the separation is in favor of o-xylene. Hu et al.48,49 report transient breakthrough experiments to demonstrate that the breakthrough of o-xylene occurs significantly later than that of either m-xylene or p-xylene, in qualitative agreement with the results presented in Fig. 22. It is also noteworthy that AlPO4-5 zeolite has been patented by Exxon Research & Engineering for o-xylene selective separation of aromatic mixtures.50
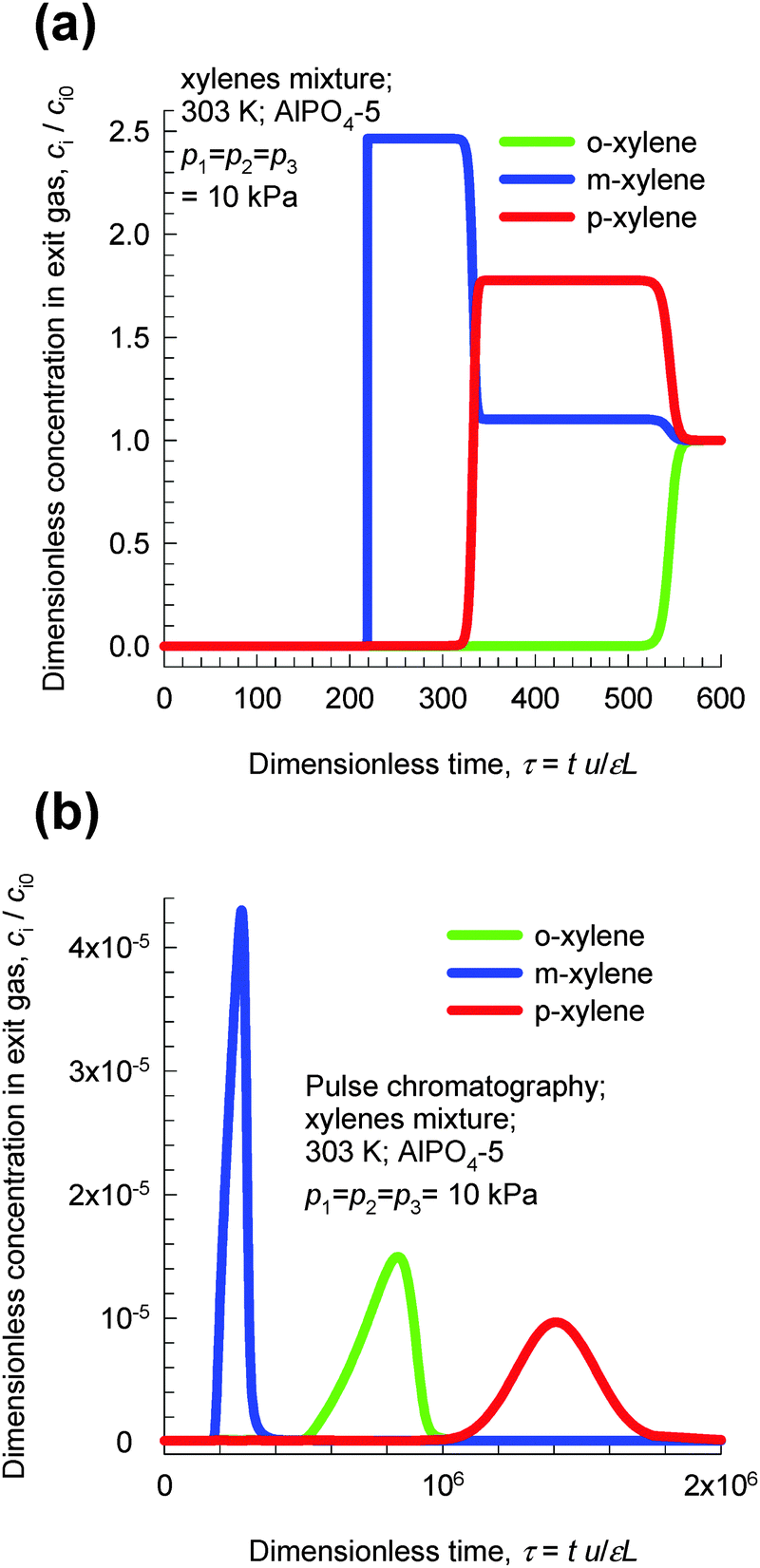 | ||
| Fig. 22 (a) Transient breakthrough simulations of step-input of 3-component o-xylene–m-xylene–p-xylene in AlPO4-5 at 303 K and a total pressure of 30 kPa. (b) Pulse chromatographic simulations of pulse-input of 3-component o-xylene–m-xylene–p-xylene in AlPO4-5 at 303 K and a total pressure of 30 kPa. The duration of the pulse is 10 s. Video animations of these transient breakthroughs are available as ESI.† | ||
Fig. 22b presents chromatographic simulations of pulse injection of an equimolar mixture at 30 kPa in a bed of AlPO4-5. We note that the elution sequence is now m-xylene, o-xylene, and p-xylene. This sequence is dictated by the adsorption strengths in the Henry regime. The point we wish to stress here is that pulse chromatographic experiments and simulations do not reflect molecular packing effects that manifest at pore saturation. It is therefore surprising to note that some journal papers51 that have examined the separation of xylene isomers with MOFs have presented pulse chromatographic experiments to demonstrate separability at pore saturation.
Torres-Knoop et al.41 have adopted a conceptual approach, using CBMC simulations, for selecting MOFs that selectively adsorb p-xylene. Within the one-dimensional 10 Å channels of MAF-X8, we have commensurate stacking of p-xylene; see snapshots in Fig. 23.41 Commensurate stacking within 1D channels of MAF-X8 results in strong selectivity in favor of p-xylene as saturation conditions are approached. Torres-Knoop et al.41 have concluded that MAF-X8 is the best MOF for separation of 4-component o-xylene–m-xylene–p-xylene–ethylbenzene mixtures, and that it is significantly better than BaX which is in current use.
 | ||
| Fig. 23 Snapshots obtained from CBMC simulations,41 showing the stacking of p-xylene within 10 Å channels of MAF-X8. | ||
In a recent paper, Mukherjee et al.52 have presented pure component adsorption isotherm data at 298 K for o-xylene, m-xylene, p-xylene, and ethylbenzene in a Zn(II)-based dynamic coordination framework, [Zn4O(L)3], where the ligand L = 4,4′-((4-(tert-butyl)-1,2-phenylene)bis(oxy))dibenzoate. The MOF structure gets transformed in such a manner as to allow optimal packing of p-xylene within the cavities. The pure component isotherm data show that the saturation capacity of p-xylene is 3 mol kg−1 which is comparable to that reported for MAF-X8 by Torres-Knoop et al.41 Remarkably, Zn4O(L)3 appears to adsorb less than about 0.5 mol kg−1 of o-xylene, m-xylene, and ethylbenzene. Unfortunately, Mukherjee et al.52 do not report IAST calculations of selectivities for Zn4O(L)3, but our estimates indicate separation selectivities in favor of p-xylene in excess of 100, significantly higher than that for BaX and MAF-X8. Further experimental work on breakthroughs, and stability tests are required in order to demonstrate the efficacy of Zn4O(L)3 for replacement of BaX in the process scheme shown in Fig. 15.
Maes et al.53 and Remy et al.54 have demonstrated that MIL-47 (V) and MIL-53 (Al) also have the potential for separation of mixtures of styrene and ethylbenzene. This separation is important in the context of replacement of the distillation column shown at the top right corner in Fig. 15. Styrene is a flat, i.e. planar, molecule. By contrast, ethylbenzene is not a flat molecule. Furthermore, the dimensions of styrene are similar to those of p-xylene (cf.Fig. 16). We may conclude that MOFs that are effective for selective adsorption of p-xylene from a mixture of C8 hydrocarbons would also be effective for selective adsorption of styrene from ethylbenzene–styrene mixtures. Based on this logic, we would not expect MIL-47 and MIL-53 to be the top candidates for this separation. Indeed, the separation selectivities obtained in the experiments of Maes et al.53 and Remy et al.54 are only about 6. A fruitful area of research would be to investigate MOFs with channel dimensions of about 10 Å that would allow optimal stacking of styrene.
10. Separating hexane isomers based on molecular footprints
Consider the adsorption of a mixture of hexane isomers nC6/3MP/22DMB within the 1D channels of AFI zeolite that has 1D channels; the CBMC mixture simulation results are presented in Fig. 24a. Near pore saturation conditions, the hierarchy of adsorption strengths is nC6 < 3MP ≪ 22DMB.55 The hierarchy of component loadings is primarily dictated by the molecular footprints (see snapshots of molecules within the 1D channel in Fig. 24b). The linear nC6 has a longer “footprint” and occupies a larger segment of the channel. 22DMB is the most compact molecule and has the smallest footprint; consequently, a greater number of di-branched isomers can be located within a given length of the channels when compared to nC6. 3MP has footprints with an intermediate character. The di-branched isomers are selectively adsorbed with AFI zeolite from the nC6/3MP/22DMB mixture.55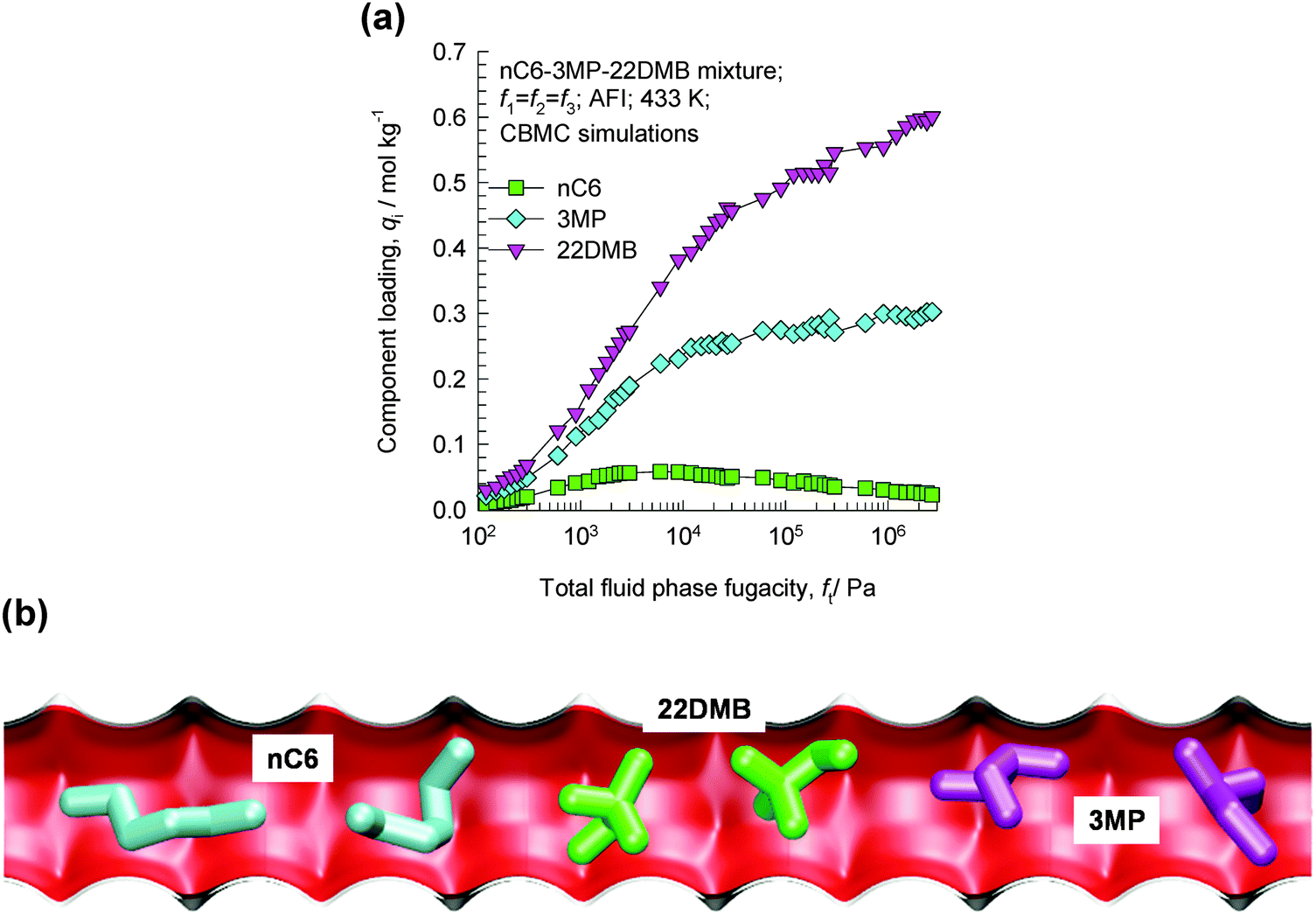 | ||
| Fig. 24 (a) CBMC simulations of adsorption equilibrium in nC6/3MP/22DMB mixtures in AFI zeolite.55 (b) Snapshots showing the molecular configurations of nC6, 3MP, and 22DMB within 1D channels of AFI zeolite.55 Video animations showing the transient breakthrough of 5-component hexane isomer mixtures in fixed beds packed with ATS and CFI are available as ESI.† | ||
ATS and CFI zeolites, patented by Chevron for application in the alkane isomerization process,56,57 have 1D channels similar to those of AFI zeolite.10,58 CBMC simulations of adsorption equilibrium in nC6/2MP/3MP/22DMB/23DMB mixtures in ATS and CFI zeolites show that linear nC6 has the lowest loading because it has the longest footprint in the 1D channels.58
From a practical point of view it is not desirable to separate hexane isomers using AFI, ATS, and CFI; we would prefer materials that selectively reject the di-branched isomers that have the highest octane numbers, as explained in previous studies.10,58
11. Separating hexane isomers based on molecular configurations
The MFI pore topology consists of a set of intersecting straight and zig-zag channels of approximately 5.5 Å size. Separations of hexane isomers with MFI are based on the exploitation of the differences in molecular configurations. The length of the n-hexane (nC6) molecule is commensurate with the distance between adjoining intersections;59 consequently, nC6 can be packed very efficiently within the channel network with a saturation loading Θi,sat = 8 molecules per unit cell; see the pure component isotherm in Fig. 25a. Branched molecules such as iso-butane (iC4), 2-methylpentane (2MP), 3-methylpentane (3MP), 2,2-dimethylbutane (22DMB), and 2,3-dimethylbutane (23DMB) are bulkier; these locate preferentially at the intersections that afford extra “leg room”. The availability of intersection sites is limited to a total of 4 per unit cell of MFI. CBMC simulations of pure component isotherms for 2MP indicate strong inflection (cf.Fig. 25a); loadings in excess of 4/uc for 2MP can only be achieved with an “extra push”, i.e. with greatly increased pressures. | ||
| Fig. 25 (a) CBMC simulations of pure component isotherms for n-hexane (nC6) and 2-methylpentane (2MP) in MFI zeolite at 298 K. (b) CBMC simulations of loadings in the adsorbed phase in equilibrium with a binary nC6/2MP mixture with partial pressures p1 = p2 in the bulk gas phase at 298 K. Also shown are IRM experimental data (open symbols).17 (c) Snapshots showing the location of nC6 (Θ1 = 2/uc) and 2MP (Θ2 = 2/uc) molecules at a total loading Θt = 4/uc. | ||
The differences in the packing efficiencies of linear and branched isomers, often referred to as configurational entropy effects,59–61 can be usefully exploited for separation of the hexane isomers to obtain different products with different degrees of branching.10,58,61,62 To illustrate this separation principle, let us consider CBMC simulations of loadings in the adsorbed phase in equilibrium with an equimolar binary gas phase containing a mixture of nC6 and 2MP with equal partial pressures p1 = p2 in the bulk gas phase; see Fig. 25b. Snapshots showing the location of nC6 and 2MP in the adsorbed phase at a total loading of 4/uc are shown in Fig. 25c. Up to total hydrocarbon pressure pt = p1 + p2 = 2 Pa, the component loading Θi of each component increases in an expected manner; increasing pt leads to a corresponding increase in the component loading Θi. At 2 Pa pressure the total loading Θt = 4/uc, signifying that all the intersection sites are fully occupied. To further adsorb 2MP we need to provide an extra “push”. Energetically, it is more efficient to obtain higher mixture loadings by “replacing” the 2MP with nC6; this configurational entropy effect is the reason behind the curious maxima in the 2MP loading in the mixture. The experimentally determined loadings from infra-red microscopy (IRM)17 are also plotted in Fig. 25b. There is good agreement of the IRM data and the CBMC mixture simulations. The IRM experiments offer direct experimental verification of the configurational entropy effects in mixture adsorption for nC6/2MP, which was first observed on the basis of CBMC mixture simulations.59,60 The data in Fig. 25b imply that sharp separations of the linear and branched hexanes are possible provided the operating conditions correspond to total mixture loadings Θt > 4/uc.
The transient uptake of nC6(1)/2MP(2) mixtures within MFI crystals was monitored using IRM in a set of four runs reported by Titze et al.17 In all four runs, the bulk gas mixture (with p1 = p2) was maintained at a constant temperature of 298 K. The total pressure pt was increased in a step-wise manner: run 1: pt = 0 to 2.6 Pa; run 2: pt = 2.6 to 4 Pa; run 3: pt = 4 to 12.2 Pa; run 4: pt = 12.2 to 102 Pa. Each run was allowed sufficient time to equilibrate, before application of the subsequent pressure step; the IRM transient uptake data are summarized in Fig. 26. We note that the transient uptake of nC6 has a fundamentally different character than that of its partner 2MP. The transient uptake of 2MP is observed to be significantly influenced by the entropy effects that govern mixture adsorption. After the initial increase in the uptake in run 1, the 2MP uptake in subsequent runs 2, 3, and 4 decreases because the total mixture loadings Θt > 4, and entropy effects are in play, causing a reduction in its equilibrium loading with the increase in pt. Entropy effects also cause the nC6 uptake to become progressively “sharper” as witnessed by the progressively faster equilibration in runs 2, 3, and 4. The continuous solid lines in Fig. 26 represent simulations of the uptake in which the Maxwell–Stefan diffusivities Đ1 and Đ2 are fitted for each of the four runs. The uptake simulations capture, nearly quantitatively, the essential features of the transient IRM uptake data for all four runs, including the nC6 overshoot observed in run 1. With increasing mixture uptake, the diffusivity of nC6 increases because the intersection sites become less blocked; this implies synergy between adsorption and diffusion.
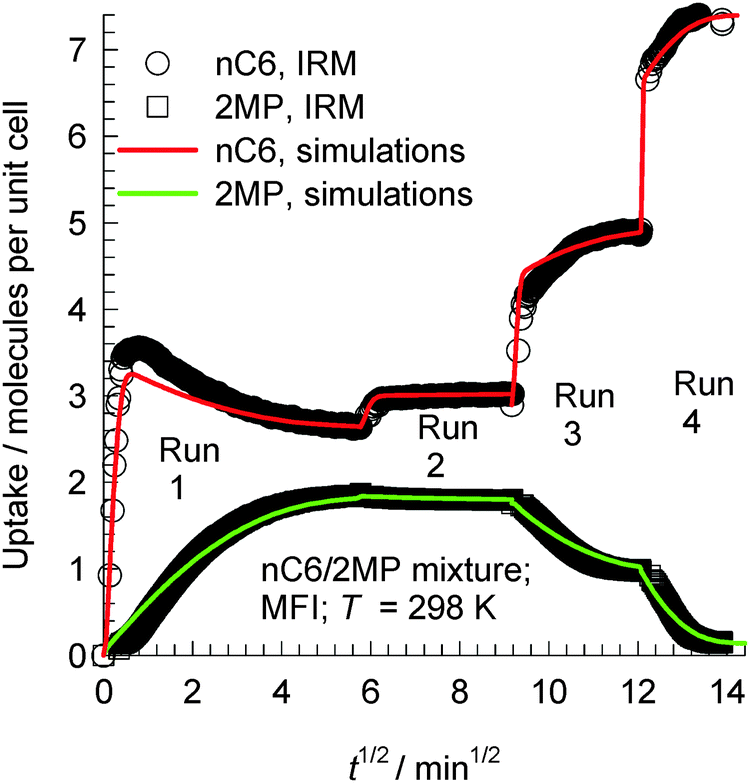 | ||
| Fig. 26 Comparison of IRM experimental data, reported by Titze et al.,17 for transient uptake of nC6/2MP mixtures with the Maxwell–Stefan model simulation results (indicated by continuous solid lines) in runs 1, 2, 3, and 4 presented sequentially. Details of the experiments and simulations are available in the paper by Titze et al.17 Video animations showing the transient breakthrough of 5-component hexane isomer mixtures in a fixed bed packed with MFI are available as ESI.† | ||
The overshoot of nC6, observed in run 1 (cf.Fig. 26), is traceable to thermodynamic coupling effects, as explained in detail in the publication of Titze et al.17 and Krishna.10 Analogous overshoots in the transient uptake of the more mobile partner species have been reported for benzene–p-xylene in ZSM-5,63,64 benzene–ethylbenzene in ZSM-5,63,64 ethanol–1-propanol in SAPO-34,65 methanol–1-propanol in SAPO-34,65 and methanol–ethanol in SAPO-34.65 Overshoots in the flux of the more mobile partner have also been reported for transient permeation of nC6/2MP,66nC6/22DMB,66m-xylene/p-xylene,67 and p-xylene/m-xylene/o-xylene67 mixtures across MFI membranes. In all the cases mentioned above, overshoots occur for the component with the higher mobility within the crystals and diffusion acts synergistically with adsorption.
The synergistic effect between adsorption and diffusion in MFI is the primary reason that MFI has superior separation performance for hexane isomer separation as compared to other zeolites and MOFs, including Fe2(BDP)3.10,58
12. Separation of C4 hydrocarbons
The separation of unsaturated olefins and dienes from C4 hydrocarbon mixtures is important in petrochemical processing. The C4 hydrocarbons possess similar sizes and boiling points. Due to the closeness of the boiling points (iso-butane = 261.45 K; iso-butene = 266.25 K; 1-butene = 266.85 K; 1,3-butadiene = 268.75 K; normal butane = 272.65 K; trans-2-butene = 273.45 K; cis-2-butene = 276.85 K), the separation of C4 streams to recover the valuable 1,3-butadiene, 1-butene, and iso-butene by distillation is difficult and energy intensive.Let us first consider the separation of n-butane (nC4) from iso-butane (iC4) using MFI zeolite that follows the same principles as for nC6/2MP mixtures considered in the foregoing section. The linear n-butane can locate anywhere along the channels. The more compact iso-butane is severely constrained within the 5.5 Å channels and prefers to locate at the intersections between the channels because such locations offer more “leg room”. The number of intersection sites is restricted to a total of 4 per unit cell of MFI. To achieve loadings of iso-butane > 4 molecules per unit cell, an “extra push” is required. Energetically, it is much more efficient to adsorb the linear n-butane to obtain higher loadings. CBMC simulations of mixture adsorption show that for total mixture loadings Θt > 4 molecules per unit cell, the selectivity is strongly in favor of the linear isomer; see Fig. 27.17,68,69 We also note that the IRM experimental data of Titze et al.17 are in excellent agreement with the CBMC mixture simulation data, and provide convincing proof of molecular packing effects that favor nC4.
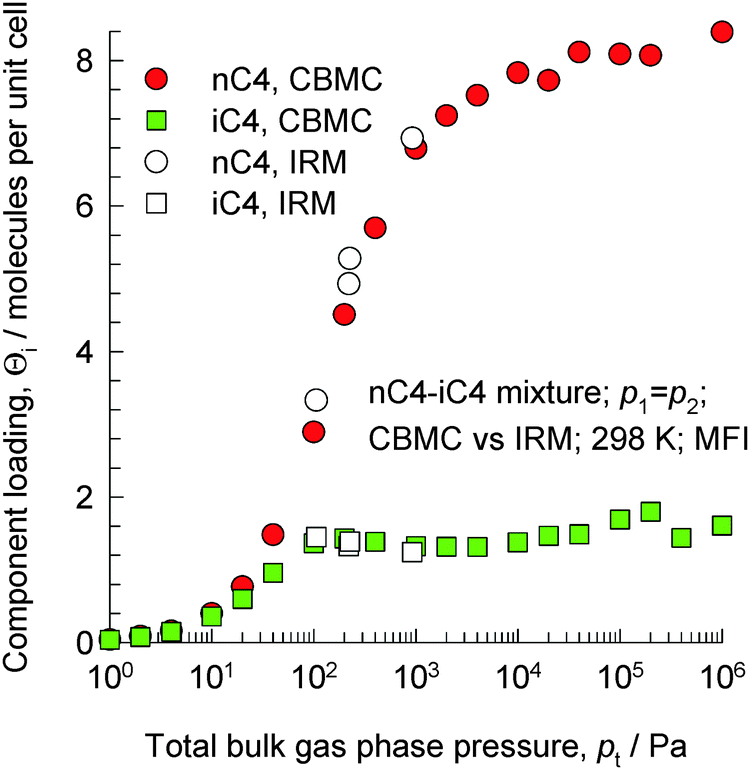 | ||
| Fig. 27 Component loadings of n-butane and iso-butane for mixture adsorption in MFI zeolite; comparison of CBMC simulations and IRM experimental data (shown by open symbols) reported by Titze et al.17 | ||
The MFI membrane permeation selectivity is found experimentally to be strongly in favor of the linear isomer.67,70 A further point to stress is that nC4/iC4 separations using MFI in both fixed bed adsorbers and membrane devices benefit from the synergy between adsorption and diffusion; both of these act in favor of the linear isomer. This has been established in the MAS PFG NMR experiments of Fernandez et al.71 and the transient uptake data of Chmelik et al.72
Tijsebaert et al.73 have demonstrated the potential of RUB-41, which has the RRO zeolite structural topology, for separation of cis-2-butene and trans-2-butene from 1-butene; this separation is of industrial importance because 1-butene is needed in the production of e.g. linear low density polyethylene (LLDPE). RUB-41 comprises a 2-dimensional intersecting channel system consisting of an 8-membered ring channel (0.27 × 0.5 nm) along [001] and a 10-membered ring channel (0.4 × 0.65 nm) along [100]. The measured data on adsorption indicate that the adsorption equilibrium and saturation capacities follow the hierarchy cis-2-butene > trans-2-butene ≫ 1-butene. This hierarchy is most likely a reflection of molecular packing effects. Breakthrough experiments of Tijsebaert et al.,73 carried out in the liquid phase, indicate that 1-butene breaks through earlier than either cis-2-butene or trans-2-butene.
Further investigations are required to find MOFs that can separate butene mixtures relying on molecular packing effects.
There are only a few studies on the separation of 1,3-butadiene from mixtures with other C4 hydrocarbons. In a US patent awarded to UOP, Preignitz74 claims the selective adsorption of 1,3-butadiene from mixtures containing other unsaturated C4 hydrocarbons using zeolite13X. The selective adsorption of 1,3-butadiene from 1-butene using Ag-Y zeolite is suggested by Takahashi et al.75 Development of MOFs for selective separation of 1,3-butadiene from C4 hydrocarbons is a fruitful area for future work.
13. Separation of chlorofluorocarbons with MFI zeolite
Let us consider the separation of CFC-115 (1-chloro-1,1,2,2,2-pentafluoroethane) and HFC-125 (1,1,1,2,2-pentafluoroethane), used as refrigerants.76 The boiling points of CFC-115 (234.1 K) and HFC-125 (224.7 K) are very close, and their mixtures can form azeotropes. Therefore, cryogenic extractive distillation has been the dominant technology utilized to separate these mixtures. Adsorptive separations offer energy-efficient alternatives to distillation. Towards this end, Peng et al.76 have reported the pure component isotherm data for CFC-115 and HFC-125 in MFI zeolite; see Fig. 28a. Due to the presence of the bulky chloro-group at the 1-position, CFC-115 locates preferentially at the intersections of MFI, which provides the required leg-room. Consequently, the pure component isotherm of CFC-115 shows a pronounced inflection at a loading of 4 molecules per unit cell. HFC-125 can locate comfortably within the channels, and the pure component isotherms do not show any pronounced inflection characteristics. | ||
| Fig. 28 (a) Pure component isotherm data of Peng et al.76 for CFC-115 and HFC-125 in MFI zeolite at 298 K. (b) IAST calculations of the component loadings for equimolar CFC-115/HFC-125 mixtures in MFI zeolite at 298 K. | ||
Fig. 28b shows IAST calculations of the component loadings for equimolar CFC-115/HFC-125 mixtures in MFI zeolite. For total pressures pt < 3 kPa, the adsorption selectivity is in favour of CFC-115. However, due to configurational entropy effects, the adsorption selectivity is in favour of HFC-125 at total pressures pt > 5 kPa. The principle of separation is entirely analogous to that of the separation of hexane isomers discussed in detail in the earlier section.
The entropy effects that cause HFC-125 to be preferentially adsorbed at high loadings are specific to MFI zeolite. An analysis of the separations of CFC-115/HFC-125 mixtures using Varuf activated carbon is presented in the ESI.† This analysis shows that CFC-115 is more strongly adsorbed in activated carbon over the entire range of pressures; there is no selectivity reversal as witnessed for MFI zeolite. Molecular packing effects are specific to guest–host combinations; they are not the exclusive property of either guest species or host material.
The development and synthesis of MOFs for separation of refrigerants is an area of research that is gaining attention in view of environmental concerns and legislations.77
14. Conclusions
The following major conclusions emerge from our detailed analysis of separations of mixtures operating close to pore saturation conditions.(1) In contrast to separations under conditions for which θt < 0.6, the separations under conditions close to pore saturation, i.e. θt ≈ 1, are significantly influenced by differences in saturation capacities. The saturation capacities are a direct reflection of the efficiencies by which molecules pack, or stack, themselves within the microporous channels. For operations in which the bulk mixture to be separated is in the liquid phase, pore saturation is assured.
(2) For mixtures of 1-alcohols, molecular packing effects can be exploited to selectively adsorb the shorter alcohol. The entropy effects are quantitatively predicted by the IAST calculations. Such separations have been experimentally demonstrated for separations with SAPO-34, ZIF-68, and UCY-5. An important feature of such separations is that intra-crystalline diffusion and mixture adsorption proceed hand-in-hand, i.e. they are synergistic.
(3) For selective adsorption of alcohols from dilute aqueous solutions, we need to use hydrophobic adsorbents in order to ensure rejection of water from the pores. This possibility has been demonstrated for ZIF-8 (cf.Fig. 7).
(4) The separation of water–benzene vapor mixtures with CuBTC is a good illustration of the relative importance of adsorption strengths and saturation capacities. For operations at lower temperatures and higher pressures, the separation is selective to water which has a higher saturation capacity. In contrast, operations at higher temperatures and lower pressures are selective to benzene which has a higher adsorption strength.
(5) Selective adsorption of water from water–alcohol mixtures can be achieved with hydrophilic adsorbents such as LTA-4A, CID-1, Cu-GLA, Co(pbdc), JUC-110, and Zn(II)-MOF by choosing conditions close to pore saturation. Large differences in saturation capacities result in better separations. In particular, CID-1 appears to have good separation potential. Another important factor is that intra-crystalline diffusion effects act in a manner that is synergistic with adsorption. Currently, LTA-4A and LTA-5A are used for selective water pervaporation in membrane processes. There is considerable scope for development of hydrophilic MOFs for water-selective separations from mixtures.
(6) The separation of o-xylene–m-xylene–p-xylene–ethylbenzene mixtures is crucially dependent on the geometry and size of the microporous channels. ortho-Xylene molecules are able to stack optimally, parallel to the 8.5 Å rhombohedric channel walls of MIL-47 and MIL-53; this results in strong selectivity to o-xylene. AlPO4-5 is also selective to o-xylene, because it has the shortest footprint within the 1D channels. MAF-X8 that has larger 10 Å channels allows efficient, commensurate, stacking of p-xylene molecules, resulting in higher saturation capacities. The MOF Zn4O(L)3 is reported by Mukherjee et al.52 to dynamically transform its crystal structure to allow optimal packing of p-xylene; it allows practically none of the other C8 aromatics to adsorb. This flexible MOF needs further experimental investigation.
(7) Pulse chromatographic experiments and simulations of the kind shown in Fig. 22b do not reflect molecular packing effects. Rather, these are dictated by adsorption strengths in the Henry regime.
(8) Separations of hexane isomers with AFI, ATS, and CFI zeolites are reliant on the differences in the footprints of linear, branched, and di-branched isomers. More compact di-branched molecules are selectively adsorbed because they have significantly shorter footprints and, consequently, higher saturation capacities. Separation based on differences in molecular footprints could be effectively utilized for separation of mixtures of butenes.73
(9) The separation of hexane isomers using MFI zeolites relies on subtle differences in molecular configurations. The linear isomers can locate comfortably along both the straight and zig-zag channels. By contrast, the mono-branched and di-branched isomers experience steric constraints and locate preferentially at the channel intersections that offer more leg-room. Since the number of intersection sites is restricted to 4 per unit cell, the saturation capacities of di-branched isomers are constrained to 4 molecules per unit cell. The differences in the packing efficiencies result in sharp separations when operating under conditions close to saturation.
(10) Mixtures of chlorofluorocarbons CFC-115 and HFC-125 can be separated using MFI zeolite by relying on the configurational differences that result from the bulky chloride at the 1-position of CFC-115. From leg-room considerations, CFC-115 prefers location at the channel intersections. Consequently, separations at high loadings become selective to HFC-115.
(11) A characteristic feature of many, if not most, separations that are reliant on molecular packing effects is that adsorption and intra-crystalline diffusion are synergistic; this enhances the separation efficiencies in fixed bed adsorbers.
Finally, we must remark that our analysis of separations in this Perspective has been conceptual. In practical applications, other aspects such as stability, regenerability, and cost will come into play in choosing the right adsorbent.
Notation
| c i | Molar concentration of species i in gas mixture, mol m−3 |
| c i0 | Molar concentration of species i in gas mixture at inlet to adsorber, mol m−3 |
| Đ i | Maxwell–Stefan diffusivity, m2 s−1 |
| f i | Partial fugacity of species i, Pa |
| f t | Total fugacity of bulk fluid mixture, Pa |
| L | Length of packed bed adsorber, m |
| p i | Partial pressure of species i in mixture, Pa |
| p t | Total system pressure, Pa |
| q i | Component molar loading of species i, mol kg−1 |
| q t | Total molar loading for mixture adsorption, mol kg−1 |
| q i,sat | Molar loading of species i at saturation, mol kg−1 |
| t | Time, s |
| T | Absolute temperature, K |
| u | Superficial gas velocity in packed bed, m s−1 |
| v | Interstitial gas velocity in packed bed, m s−1 |
Greek letters
| ε | Voidage of packed bed, dimensionless |
| θ t | Fractional occupancy for mixture adsorption, dimensionless |
| Θ i | Loading of species i, molecules per unit cage or per unit cell |
| Θ t | Total mixture loading, molecules per unit cage or per unit cell |
| τ | Time, dimensionless |
Subscripts
| i | Referring to component i |
| t | Referring to total mixture |
References
- L. Hamon, E. Jolimaître and G. D. Pirngruber, Ind. Eng. Chem. Res., 2010, 49, 7497–7503 CrossRef CAS
.
- H. Wu, K. Yao, Y. Zhu, B. Li, Z. Shi, R. Krishna and J. Li, J. Phys. Chem. C, 2012, 116, 16609–16618 CAS
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030–3040 CAS
- Z. R. Herm, J. A. Swisher, B. Smit, R. Krishna and J. R. Long, J. Am. Chem. Soc., 2011, 133, 5664–5667 CrossRef CAS PubMed
- Y. Belmabkhout, G. Pirngruber, E. Jolimaitre and A. Methivier, Adsorption, 2007, 13, 341–349 CrossRef CAS PubMed
- S. U. Rege and R. T. Yang, Ind. Eng. Chem. Res., 1997, 36, 5358–5365 CrossRef CAS
- J. Liu, P. K. Thallapally and D. Strachan, Langmuir, 2012, 28, 11584–11589 CrossRef CAS PubMed
- J. Liu, D. M. Strachan and P. K. Thallapally, Chem. Commun., 2014, 50, 466–468 RSC
- Y. Gurdal and S. Keskin, Ind. Eng. Chem. Res., 2012, 51, 7373–8382 CrossRef CAS
- R. Krishna, Microporous Mesoporous Mater., 2014, 185, 30–50 CrossRef CAS PubMed
- J. C. Saint-Remi, T. Rémy, V. van Huskeren, S. van de Perre, T. Duerinck, M. Maes, D. E. De Vos, E. Gobechiya, C. E. A. Kirschhock, G. V. Baron and J. F. M. Denayer, ChemSusChem, 2011, 4, 1074–1077 CrossRef PubMed
- M. Minceva and A. E. Rodrigues, AIChE J., 2007, 53, 138–149 CrossRef CAS
- M. Minceva and A. E. Rodrigues, Chem. Eng. Res. Des., 2004, 82, 667–681 CrossRef CAS
- R. Krishna and J. M. van Baten, Sep. Purif. Technol., 2011, 76, 325–330 CrossRef CAS PubMed
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–130 CrossRef CAS
- T. Remy, J. C. Saint-Remi, R. Singh, P. A. Webley, G. V. Baron and J. F. M. Denayer, J. Phys. Chem. C, 2011, 115, 8117–8125 CAS
- T. Titze, C. Chmelik, J. Kärger, J. M. van Baten and R. Krishna, J. Phys. Chem. C, 2014, 116, 2660–2665 Search PubMed
- R. Krishna and J. M. van Baten, Sep. Purif. Technol., 2008, 60, 315–320 CrossRef CAS PubMed
- K. Zhang, R. P. Lively, M. E. Dose, A. J. Brown, C. Zhang, J. Chung, S. Nair, W. J. Koros and R. R. Chance, Chem. Commun., 2013, 49, 3245–3247 RSC
- S. Van der Perre, T. Van Assche, B. Bozbiyik, J. Lannoeye, D. E. De Vos, G. V. Baron and J. F. M. Denayer, Langmuir, 2014, 30, 8416–8424 CrossRef CAS PubMed
- C. Chmelik, J. Kärger, M. Wiebcke, J. Caro, J. M. van Baten and R. Krishna, Microporous Mesoporous Mater., 2009, 117, 22–32 CrossRef CAS PubMed
- Z. Zhao, S. Wang, Y. Yang, X. Li, J. Li and Z. Li, Chem. Eng. J., 2015, 259, 79–89 CrossRef CAS PubMed
- A. Nalaparaju, X. S. Zhao and J. W. Jiang, J. Phys. Chem. C, 2010, 114, 11542–11550 CAS
- R. Krishna and J. M. van Baten, Langmuir, 2010, 26, 10854–10867 CrossRef CAS PubMed
- J. J. Pluth and J. V. Smith, J. Am. Chem. Soc., 1980, 102, 4704–4708 CrossRef CAS
- A. García-Sánchez, E. García-Pérez, D. Dubbeldam, R. Krishna and S. Calero, Adsorpt. Sci. Technol., 2007, 25, 417–427 CrossRef
- N. Hedin, G. J. DeMartin, W. J. Roth, K. G. Strohmaier and S. C. Reyes, Microporous Mesoporous Mater., 2008, 109, 327–334 CrossRef CAS PubMed
- N. Hedin, G. J. DeMartin, K. G. Strohmaier and S. C. Reyes, Microporous Mesoporous Mater., 2007, 98, 182–188 CrossRef CAS PubMed
- S. Fritzsche, R. Haberlandt, J. Kärger, H. Pfeifer, K. Heinzinger and M. Wolfsberg, Chem. Phys. Lett., 1995, 242, 361–366 CrossRef CAS
- M. Simo, S. Sivashanmugam, C. J. Brown and V. Hlavacek, Ind. Eng. Chem. Res., 2009, 48, 9257–9260 CrossRef
- C. G. Efthymiou, E. J. Kyprianidou, C. J. Milios, M. J. Manos and A. J. Tasiopoulos, J. Mater. Chem. A, 2013, 1, 5061–5069 CAS
- Z. Lin, R. Zou, J. Liang, W. Xia, D. Xia, Y. Wang, J. Lin, T. Hu, Q. Chen, X. Wang, Y. Zhao and A. K. Burrell, J. Mater. Chem., 2012, 22, 7813–7818 RSC
- X. Zheng, L. Zhou, Y. Huang, C. Wang, J. Duan, L. Wen, Z. Tian and D. Li, J. Mater. Chem. A, 2014, 2, 12413–12422 CAS
- S. Horike, D. Tanaka, K. Nakagawa and S. Kitagawa, Chem. Commun., 2007, 3395–3397 RSC
- B. Chen, Y. Ji, M. Xue, F. R. Fronczek, E. J. Hurtado, J. U. Mondal, C. Liang and S. Dai, Inorg. Chem., 2008, 47, 5543–5545 CrossRef CAS PubMed
- X. Zheng, Y. Huang, J. Duan, C. Wang, L. Wen, J. Zhao and D. Li, Dalton Trans., 2014, 43, 8311–8317 RSC
- J.-Z. Gu, W.-G. Lu, L. Jiang, H. C. Zhou and T.-B. Lu, Inorg. Chem., 2007, 46, 5835–5837 CrossRef CAS PubMed
- T. Borjigin, F. Sun, J. Zhang, K. Cai, H. Ren and G. S. Zhu, Chem. Commun., 2012, 48, 7613–7615 RSC
- M. Minceva and A. E. Rodrigues, Ind. Eng. Chem. Res., 2002, 41, 3454–3461 CrossRef CAS
- D. Peralta, K. Barthelet, J. Pérez-Pellitero, C. Chizallet, G. Chaplais, A. Simon-Masseron and G. D. Pirngruber, J. Phys. Chem. C, 2012, 116, 21844–21855 CAS
- A. Torres-Knoop, R. Krishna and D. Dubbeldam, Angew. Chem., Int. Ed., 2014, 53, 7774–7778 CrossRef CAS PubMed
- R. El Osta, A. Carlin-Sinclair, N. Guillou, R. I. Walton, F. Vermoortele, M. Maes, D. De Vos and F. Millange, Chem. Mater., 2012, 24, 2781–2791 CrossRef CAS
- V. Finsy, H. Verelst, L. Alaerts, D. De Vos, P. A. Jacobs, G. V. Baron and J. F. M. Denayer, J. Am. Chem. Soc., 2008, 130, 7110–7118 CrossRef CAS PubMed
- T. Remy, G. V. Baron and J. F. M. Denayer, Langmuir, 2011, 27, 13064–13071 CrossRef CAS PubMed
- F. Niekiel, J. Lannoeye, H. Reinsch, A. S. Munn, A. Heerwig, I. Zizak, S. Kaskel, R. I. Walton, D. de Vos, P. Llewellyn, A. Lieb, G. Maurin and N. Stock, Inorg. Chem., 2012, 53, 4610–4620 CrossRef PubMed
- Z. L. Fang, S. R. Zheng, J. B. Tan, S. L. Cai, J. Fan, X. Yan and W. G. Zhang, J. Chromatogr. A, 2013, 1285, 132–138 CrossRef CAS PubMed
- A. S. T. Chiang, C.-K. Lee and Z.-H. Chang, Zeolites, 1991, 380–386 CrossRef CAS
- E. Hu, A. T. Derebe, A. Almansoori and K. Wang, Int. J. Mater. Sci. Eng., 2014, 2, 10–14 Search PubMed
- E. Hu, Z. Lai and K. Wang, J. Chem. Eng. Data, 2010, 55, 3286–3289 CrossRef CAS
-
D. D. Rosenfeld and D. M. Barthomeuf, Separation of Ortho Aromatic Isomers by Selective Adsorption with an Aluminophosphate, US Pat., US 4482776, Exxon Research & Engineering Co., Florham Park, N.J., 1984 Search PubMed
- L. Alaerts, C. E. A. Kirschhock, M. Maes, M. van der Veen, V. Finsy, A. Depla, J. A. Martens, G. V. Baron, P. A. Jacobs, J. F. M. Denayer and D. De Vos, Angew. Chem., Int. Ed., 2007, 46, 4293–4297 CrossRef CAS PubMed
- S. Mukherjee, B. Joarder, B. Manna, A. V. Desai, A. K. Chaudhari and S. K. Ghosh, Sci. Rep., 2014, 4, 5761, DOI:10.1038/srep05761
- M. Maes, F. Vermoortele, L. Alaerts, S. Couck, C. E. A. Kirschhock, J. F. M. Denayer and D. E. De Vos, J. Am. Chem. Soc., 2010, 132, 15277–15285 CrossRef CAS PubMed
- T. Remy, L. Ma, M. Maes, D. E. De Vos, G. V. Baron and J. F. M. Denayer, Ind. Eng. Chem. Res., 2012, 51, 14824–14833 CrossRef CAS
- R. Krishna and J. M. van Baten, Phys. Chem. Chem. Phys., 2011, 13, 10593–10616 RSC
-
T. Maesen and T. Harris, Process for producing high RON gasoline using CFI zeolite, US Pat., US 70374222 B2, Chevron U.S.A. Inc, San Ramon, CA, US, 2006 Search PubMed
-
T. Maesen and T. Harris, Process for producing high RON gasoline using ATS zeolite, US Pat., US 7029572 B2, Chevron U.S.A. Inc, San Ramon, CA, US, 2006 Search PubMed
- Z. R. Herm, B. M. Wiers, J. M. Van Baten, M. R. Hudson, P. Zajdel, C. M. Brown, N. Maschiocchi, R. Krishna and J. R. Long, Science, 2013, 340, 960–964 CrossRef CAS PubMed
- T. J. H. Vlugt, R. Krishna and B. Smit, J. Phys. Chem. B, 1999, 103, 1102–1118 CrossRef CAS
- R. Krishna, B. Smit and T. J. H. Vlugt, J. Phys. Chem. A, 1998, 102, 7727–7730 CrossRef CAS
- R. Krishna, Chem. Eng. Res. Des., 2001, 79, 182–194 CrossRef CAS
- D. Dubbeldam, R. Krishna, S. Calero and A. Ö. Yazaydın, Angew. Chem., Int. Ed., 2012, 51, 11867–11871 CrossRef CAS PubMed
- W. Niessen and H. G. Karge, Microporous Mater., 1993, 1, 1–8 CrossRef CAS
- H. G. Karge, C. R. Chim., 2005, 8, 303–319 CrossRef CAS PubMed
- J. C. Saint-Remi, G. V. Baron and J. F. M. Denayer, J. Phys. Chem. C, 2013, 117, 9758–9765 Search PubMed
- T. Matsufuji, K. Watanabe, N. Nishiyama, Y. Egashira, M. Matsukata and K. Ueyama, Ind. Eng. Chem. Res., 2000, 39, 2434–2438 CrossRef CAS
- T. Matsufuji, N. Nishiyama, M. Matsukata and K. Ueyama, J. Membr. Sci., 2000, 178, 25–34 CrossRef CAS
- R. Krishna and D. Paschek, Sep. Purif. Technol., 2000, 21, 111–136 CrossRef CAS
- R. Krishna and D. Paschek, Phys. Chem. Chem. Phys., 2001, 3, 453–462 RSC
- Z. A. E. P. Vroon, K. Keizer, M. J. Gilde, H. Verweij and A. J. Burggraaf, J. Membr. Sci., 1996, 113, 293–300 CrossRef CAS
- M. Fernandez, J. Kärger, D. Freude, A. Pampel, J. M. van Baten and R. Krishna, Microporous Mesoporous Mater., 2007, 105, 124–131 CrossRef CAS PubMed
- C. Chmelik, L. Heinke, J. M. van Baten and R. Krishna, Microporous Mesoporous Mater., 2009, 125, 11–16 CrossRef CAS PubMed
- B. Tijsebaert, C. Varszegi, H. Gies, F. S. Xiao, X. Bao, T. Tatsumi, U. Müller and D. De Vos, Chem. Commun., 2008, 2480–2482 RSC
-
J. W. Priegnitz, Process for the Separation of 1,3-butadient by Selective Adsorption on a Zeolite Adsorbent, US Pat., US 3992471, UOP Inc., Des Plaines, Illinois, 1976 Search PubMed
- A. Takahashi, R. T. Yang, C. L. Munson and D. Chinn, Ind. Eng. Chem. Res., 2001, 40, 3979–3988 CrossRef CAS
- Y. Peng, Z. Zhang, X. Zheng, H. Wang, C. Xu, Q. Xiao, Y. Zhong and W. Zhu, Ind. Eng. Chem. Res., 2010, 49, 10009–10015 CrossRef CAS
- R. K. Motkuri, H. V. R. Annapureddy, M. Vijaykumar, T. Schaef, P. F. Martin, B. P. McGrail, L. X. Dang, R. Krishna and P. K. Thallapally, Nat. Commun., 2014, 5, 4368, DOI:10.1038/ncomms5368
Footnote |
| † Electronic supplementary information (ESI) available: This material provides (a) structural information on zeolites, MOFs, and ZIFs, (b) CBMC simulation methodology, (c) simulation methodology for transient breakthroughs, (d) data on pure component isotherms used to analyze the variety of separations, (e) IAST calculations of mixture adsorption equilibria, (f) details of simulations of transient breakthroughs of mixtures in fixed bed adsorbers, (g) additional background information of the importance of MOFs in hydrocarbon separations. Also included as ESI are video animations for transient breakthroughs. See DOI: 10.1039/c4cp03939d |
| This journal is © the Owner Societies 2015 |