
Oxidation behavior of P3HT layers on bare and TiO2-covered ZnO ripple structures evaluated by photoelectron spectroscopy†
Dae Han
Kim
,
Myung-Geun
Jeong
,
Hyun Ook
Seo
and
Young Dok
Kim
*
Department of Chemistry, Sungkyunkwan University, Suwon, 440-746, Korea. E-mail: ydkim91@skku.edu; Fax: +82 31 290 7075; Tel: +82 31 299 4564
First published on 7th November 2014
Abstract
P3HT layers with a thickness of ∼5 nm were deposited on bare and TiO2-covered ZnO ripple structures. The ZnO ripples were prepared wet-chemically and a TiO2 layer with a thickness less than 5 nm was prepared by atomic layer deposition. Under humid air and visible light illumination, the oxidation behaviors of P3HT on these surfaces were studied using photoelectron spectroscopy. It was found that P3HT on TiO2/ZnO oxidizes more easily than that on bare ZnO ripples. Using a model substrate of a flat ZnO surface in combination with angle-resolved photoelectron spectroscopy, we found that oxidation of P3HT occurs at the surface of the topmost layer of P3HT, not at the P3HT/oxide interfaces, even though P3HT oxidation is strongly influenced by the interface structure. It is suggested that the lifetime of electron–hole pairs can be strongly influenced by the interface structure, which can also affect the oxidation behavior of P3HT.
1. Introduction
Polythiophene derivatives, particularly poly(3-hexylthiophene-2,5-diyl) (P3HT), have been widely used in organic electronics such as organic light emitting diodes1,2 and organic solar cells.3–5 The low stability of these polymers, which is due to easy oxidation of the polymers under light illumination, is a hurdle which must be overcome for practical use of organic electronics.6–8 This problem is particularly important for the development of organic solar cells since the first step of the operation of organic solar cells is absorption of light by an organic polymer, which can create electricity and also initiate photo-induced degradation of the organic polymers. Much effort has been previously devoted to the characterization of the degradation behavior of organic solar cells by monitoring the change in the device performance as a function of the light illumination time under air. More recently, spectroscopic methods have been employed to investigate the oxidation mechanisms of organic polymers in solar cells.9–15 It was suggested that the initial decrease of the power conversion efficiency of organic solar cells is related to the oxidation of the organic polymer at polymer/oxide interfaces,16,17 whereas slower degradation was found to be related to the reaction between the polymer and oxygen/water present in the atmosphere.10–13 Oxidation of the polymer was found to be strongly influenced by the types of buffer layers in the organic solar cell.18–21 In the case of organic solar cells containing buffer layers consisting of inorganic materials (e.g., TiO2), oxidation of the polymer was suggested to be initiated by UV absorption of inorganic materials, which can result in the formation of electron–hole pairs and subsequently, strong oxidizing agents such as O2− and OH radicals due to interactions between electron–hole pairs and oxygen/water.20–22 These oxidizing agents can react with organic polymers, leading to the degradation of the polymers.In this study, we focused on the oxidation of a P3HT layer on ZnO ripple structures with and without additional TiO2 layers on ZnO. It was previously observed that the ZnO ripple structure as an electron-accepting layer of an organic solar cell can demonstrate an enhanced power conversion efficiency compared to a flat ZnO layer.23 With additional TiO2 thin layers with thicknesses less than 5 nm prepared by atomic layer deposition (ALD), we obtained an even higher organic solar cell performance than that of bare ZnO ripples. It was concluded that the ALD-TiO2 layer can reduce the number of defect sites of wet-chemically prepared ZnO ripples, which can act as recombination centers of electron–hole pairs and thus, reduce the photocurrent.24
In the present work, photoelectron spectroscopy was employed to investigate the initial oxidation behaviors of P3HT on bare and TiO2-covered ZnO ripples in well-defined oxidizing environments. We used visible light illumination, avoiding absorption of light by the oxide buffer layer, and having optical excitation only at the organic polymer layer.
2. Experimental details
2.1. ALD for thin film preparation
For deposition of TiO2 thin films using the ALD method on ZnO ripples, titanium tetraisopropoxide (TTIP, Aldrich) and distilled H2O were used as precursors, which were sequentially introduced into the vacuum chamber with a base pressure of ∼2 mTorr. For each ALD cycle, a TTIP pulse of 10 s with a working pressure of ∼300 mTorr was followed by a purging step, and N2 (99.999%) was used as a purging gas. Then, a H2O pulse of 10 s with a working pressure of ∼600 mTorr and subsequent purging was used. Substrate temperature during deposition was 150 °C, and TTIP and H2O bottle temperatures were 60 °C and room temperature, respectively.A ZnO film was deposited on indium tin oxide (ITO) using the ALD method. Diethylzinc (DEZ, UPChem) and distilled H2O were used as zinc and oxygen precursors, respectively. For each ALD cycle, DEZ and H2O were injected for 10 and 5 s, respectively into the vacuum chamber. During ZnO deposition, the base pressure was kept at ∼1 mTorr by purging and pumping steps. Both DEZ and H2O bottles were maintained at room temperature, and the substrate was maintained at 130 °C.
2.2. Wet-chemical preparation
In order to prepare a ZnO nano-ripple film, 0.75 M ZnO sol–gel solution was spin-coated onto ITO-coated glass pretreated with UV-ozone for 30 min. The solution was prepared by dissolving zinc acetate [Zn(CH3COO)2·2H2O] in a 2-methoxyethanol solvent containing ethanolamine as a stabilizer. After spin-coating, the sample was heated to 350 °C with a constant heating rate of 22 °C min−1 in a furnace.The P3HT thin layer was also deposited on the fabricated ZnO ripple surfaces using a spin-coating technique. P3HT (regioregularity ∼94%, 4002-EE, Rieke Metals, Inc.) was used without further purification; 20 mg of regioregular P3HT was dissolved into 1 ml of 1,2-dichlorobenzene (DCB, Aldrich) at 60 °C and stirred overnight. A thin P3HT layer with a thickness of ∼5 nm was spin-coated with 20 μl of the P3HT solution at 3500 rpm for 40 s.
2.3. In situ X-ray photoelectron spectroscopy (XPS)
The preparation (base pressure ∼7.0 × 10−8 Torr) and main (base pressure ∼3.0 × 10−10 Torr) chambers were connected via a gate valve allowing the sample to be transferred between these two chambers without exposure to air.25 XPS spectra (S 2p and C 1s core-level) of the as-prepared P3HT/ZnO ripple sample were repeatedly obtained after back-and-forth transfer between the preparation and main chambers. After each gas exposure in the preparation chamber under light illumination, the sample surface was analyzed in the main chamber which was equipped with a dual Al/Mg X-ray source and a concentric hemispherical analyzer (CHA, PHOIBOS-Has 2500, SPECS). XPS spectra were obtained at room temperature using a Mg-Kα (1253.6 eV) X-ray source.Gas exposure experiments were performed under light illumination using a blue light emitting diode (LED) with a maximum power of 700 W m−2 at 455 (±40) nm in the preparation chamber.11 The absorption of light by ZnO and TiO2 in our experiment is negligible, since there is almost no overlap between the light emission profile of our blue LED and the optical absorption of ZnO and TiO2.24 In contrast, P3HT has a very high absorption coefficient of light at the wavelength range of our light source. The chamber was composed of a quartz window which was used to allow light exposure to the sample from an external source. The sample was exposed to an atmosphere of humid air (dry air and water vapor with 20% of relative humidity) at room temperature.
3. Results and discussion
Fig. 1a shows S 2p spectra collected from P3HT layers deposited on rippled ZnO surfaces before and after exposure to a humid atmosphere under blue LED illumination for various exposure times. For the as-prepared sample, a doublet structure with a peak area ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was obtained and these two peaks can be assigned to the 2p3/2 and 2p1/2 states of S, respectively. The 2p3/2 state centered at 164.7 eV can be attributed to the non-oxidized S species of P3HT and an additional doublet structure with its 2p3/2 centered at 166.7 eV is due to sulfone species (O
:1 was obtained and these two peaks can be assigned to the 2p3/2 and 2p1/2 states of S, respectively. The 2p3/2 state centered at 164.7 eV can be attributed to the non-oxidized S species of P3HT and an additional doublet structure with its 2p3/2 centered at 166.7 eV is due to sulfone species (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO).10,11,26 As shown in Table 1, the relative amount of sulfone with respect to the non-oxidized S in the as-prepared sample was determined to be negligibly small and the existence of sulfone species in the as-prepared sample seems to be due to trace oxygen impurities existing in the sample. With increasing exposure time to the humid atmosphere with light illumination, growth of a new S 2p state with a 2p3/2 binding energy of 165.2 eV was observed, which can be assigned to the formation of sulfoxide species (SO).10,11,26 No additional state appeared at binding energies above 168.0 eV, indicating that the conjugated ring structure of P3HT was mostly maintained during humid air exposure for 18 h.10,13,26,27
SO).10,11,26 As shown in Table 1, the relative amount of sulfone with respect to the non-oxidized S in the as-prepared sample was determined to be negligibly small and the existence of sulfone species in the as-prepared sample seems to be due to trace oxygen impurities existing in the sample. With increasing exposure time to the humid atmosphere with light illumination, growth of a new S 2p state with a 2p3/2 binding energy of 165.2 eV was observed, which can be assigned to the formation of sulfoxide species (SO).10,11,26 No additional state appeared at binding energies above 168.0 eV, indicating that the conjugated ring structure of P3HT was mostly maintained during humid air exposure for 18 h.10,13,26,27
 | ||
| Fig. 1 S 2p spectra collected before and after exposure to humid air conditions with visible light illumination: (a) P3HT layer on bare ZnO ripple surfaces and (b) P3HT layer on ZnO ripple surfaces covered with an ALD-deposited TiO2 layer. | ||
| Non-oxidizeda (164.7 eV) | Sulfoxidea (165.2 eV) | Sulfonea (166.7 eV) | Total-oxidizeda (∼170.0 eV) | ||
|---|---|---|---|---|---|
| Unit: %.a Binding energy of the 2p3/2 state of S. | |||||
| ZnO ripple | As prepared | 93.3 | 0.0 | 6.3 | 0.4 |
| 3 h | 86.5 | 6.4 | 6.0 | 1.1 | |
| 12 h | 77.0 | 17.2 | 4.8 | 1.0 | |
| 18 h | 61.2 | 31.8 | 4.0 | 3.0 | |
| TiO2/ZnO ripple | As prepared | 93.3 | 0.0 | 6.1 | 0.6 |
| 3 h | 80.7 | 12.5 | 5.6 | 1.2 | |
| 12 h | 24.2 | 64.9 | 5.4 | 5.5 | |
| 18 h | 21.3 | 62.8 | 6.0 | 9.9 | |
Fig. 1b shows the results of similar experiments using TiO2-layered ZnO ripples as the substrate of the P3HT layers. A 5 nm thick TiO2 film on ZnO was prepared by ALD and we recently showed that such an ALD-prepared TiO2 film can form a conformal and homogeneous layer on ZnO ripples without altering the surface morphologies of the rippled structure.28,29 As seen in Fig. 1 and Table 1, sulfoxide formation in the P3HT upon humid air exposure with visible light illumination on TiO2/ZnO is much more pronounced than on bare ZnO. It is also notable that S 2p3/2 peaks centered at 169.0–170.0 eV can only be observed in the presence of the TiO2 layer, which can be attributed to the oxidized S species with ring-opening (e.g. sulfate).13,27,30
Fig. 2 and Table 2 show the results of C 1s spectra collected from the same samples shown in Fig. 1. For the as-prepared P3HT/ZnO, a single pronounced peak centered at ∼286.0 eV can be observed and careful analysis based on the literature reveals that the C 1s peak can be de-convoluted into six different components.25,27,30,31 The three states centered at 285.3, 285.9, and 286.1 eV can be attributed to CC–C, C–C, and CC–S groups, respectively, with a relative intensity of 1:3:1, corresponding to the molecular structure of P3HT. The peaks at 286.9, 288.3, and 289.8 eV can be attributed to various oxidized species of the C of P3HT (C–OH, CO, and COOH, respectively). With increasing exposure time to humid air, the relative amount of oxidized species of C increases. Particularly, the increase of the intensity of the C–OH component is much more pronounced than the other oxidized C species. In a previous work, C atoms in the α-position of P3HT have been suggested to be preferentially oxidized to C–OH.12 The results of the C 1s spectra are in line with those of the S 2p spectra where C atoms in P3HT on TiO2/ZnO are more oxidized than on bare ZnO. In the case of bare ZnO, the oxidation of C mostly resulted in the production of C–OH (Fig. 2a). However, the formation of C–OH was accompanied by the appearance of the CO peak in the case of oxidation of P3HT on TiO2/ZnO (Fig. 2b).
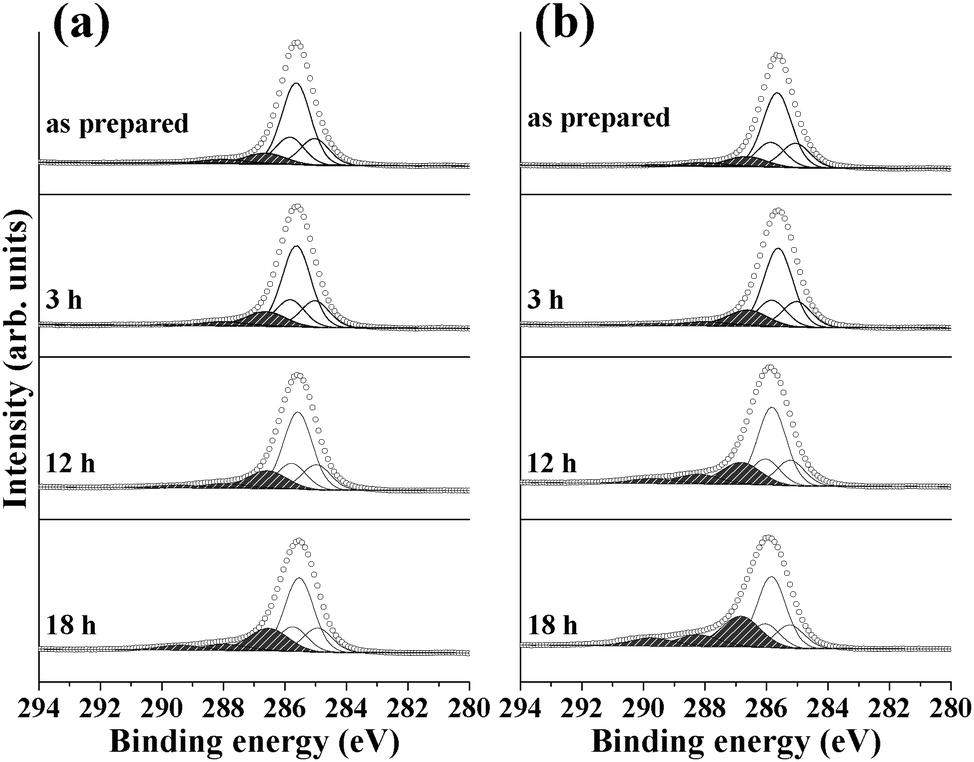 | ||
| Fig. 2 C 1s spectra collected before and after exposure to humid air conditions with visible light illumination: (a) P3HT layer on bare ZnO ripple surfaces and (b) P3HT layer on ZnO ripple surfaces covered with an ALD-deposited TiO2 layer. | ||
| C–C (285.9 eV) | CC–C (285.3 eV) |
CC–S (286.1 eV) |
C–OH (286.9 eV) | CO (288.3 eV) |
COOH (289.8 eV) | ||
|---|---|---|---|---|---|---|---|
| Unit: %. | |||||||
| ZnO ripple | As prepared | 52.5 | 17.3 | 17.3 | 8.9 | 3.2 | 0.8 |
| 3 h | 50.4 | 16.7 | 16.6 | 11.8 | 3.1 | 1.4 | |
| 12 h | 48.1 | 15.9 | 15.9 | 14.6 | 3.4 | 2.1 | |
| 18 h | 44.4 | 14.6 | 14.6 | 18.0 | 4.6 | 3.8 | |
| TiO2/ZnO ripple | As prepared | 52.2 | 17.3 | 17.3 | 9.1 | 3.1 | 1.0 |
| 3 h | 49.9 | 16.4 | 16.4 | 13.2 | 2.7 | 1.4 | |
| 12 h | 43.9 | 14.5 | 14.5 | 16.4 | 7.0 | 3.7 | |
| 18 h | 38.7 | 12.7 | 12.7 | 22.2 | 8.1 | 5.6 | |
Fig. 3 displays the valence band XPS and O 1s core-level spectra obtained from the data shown in Fig. 1 and 2. For both the valence band and O 1s spectra, it is obvious that the structural changes in the polymer layer on TiO2/ZnO upon exposure to air and light are more pronounced than those in the bare ZnO ripples. In the valence band spectra of bare P3HT layers on both substrates (Fig. 3a and c), a feature centered at 4.5 eV is observed along with a broader state at 6.0–8.0 eV. These two features at lower and higher binding energies can be attributed to the π- and σ-states of P3HT, respectively.32 With increasing exposure time to humid air and light, the π-derived states do not show much change, whereas positive shifts can be found for the σ-states, indicating that oxidation of P3HT occurs with a sustained ring structure. This result is in line with the S 2p data, in which the formation of sulfone and sulfoxide species with almost no S species causing ring-opening was observed upon oxidation. For bare ZnO, only a positive shift of the σ-states by exposure to humid air and light can be observed, whereas that of TiO2/ZnO not only shows a positive shift but also decreases of the intensities of the σ-states with increasing exposure time. The intensity of the O 1s spectrum increases with increasing humid air and light exposure times, in line with our observation of the positive core-level shifts of S and C upon the same treatments (Fig. 3b and d). Here, the increase of the O 1s intensity of TiO2/ZnO is also more pronounced than that of bare ZnO. The O 1s peak appearing after oxidation is centered at 533.0 eV and a shoulder at 535.0 eV also appears. The peak at 533.0 eV can be attributed to OH or O species bound to C and S, and the peak at 535.0 eV corresponds to molecular water.30 We previously confirmed that partial oxidation of P3HT induces the formation of conformational defects and water molecules can be incorporated into the P3HT layer via conformational defects.11
 | ||
| Fig. 3 ((a) and (c)) Valence band XPS spectra and ((b) and (d)) O 1s spectra of the samples shown in Fig. 1 and 2. (a) and (b) were obtained from bare ZnO ripples, whereas (c) and (d) were collected from TiO2-deposited ZnO ripples. | ||
From the comparison of the data of TiO2/ZnO and bare ZnO, one can conclude that the oxidation behavior of P3HT is significantly influenced by the structure of the substrate. Considering that TiO2 layers on ZnO deposited by ALD form a very thin and conformal layer with only minor alteration of the surface morphology,28,29 different electronic structures of both substrates should be considered to account for the dissimilar oxidation behaviors of P3HT on the substrates. Considering that P3HT oxidation is very much dependent on the structure of the oxide substrate, a question can be raised whether oxidation of P3HT preferentially takes place at P3HT/oxide interfaces or not. In order to shed light on this question, we evaluated oxidation of P3HT on flat ALD-deposited ZnO layers using angle-resolved XPS. The use of angle-resolved XPS for depth analysis is limited when the substrate has a rough structure.
Fig. 4a shows the S 2p spectrum of the P3HT layer on an ALD-ZnO film, which is deposited on an indium tin oxide (ITO) substrate. Similar to the results shown in Fig. 1, exposure of the sample to humid air and light causes sulfoxide formation. Only minor increases of the core-level peak intensity can be observed for S species with further oxidation (e.g., sulfone and sulfate). Fig. 4b shows the S 2p spectrum of the oxidized P3HT/ALD-ZnO sample shown in Fig. 4a taken at a different aperture angle of the analyzer normal to the surface of the sample (0° in Fig. 4a and 60° in Fig. 4b). The spectrum in Fig. 4b should be more surface sensitive than that shown in Fig. 4a.33 Compared to the spectrum shown in Fig. 4a, which had lower surface sensitivity, oxidation of the P3HT layer is much more pronounced in Fig. 4b where the increase of the intensity of the sulfoxide species with increasing exposure time is much faster. In addition, highly oxidized S species with an open ring-structure at binding energies higher than 169.0 eV can be more clearly seen in the spectrum with a higher surface sensitivity. This result clearly shows that surface topmost layers of P3HT are preferentially oxidized, not the P3HT buried at the P3HT/oxide interfaces.
 | ||
| Fig. 4 P3HT oxidation on ALD-ZnO/ITO was studied under the same conditions as the samples shown in Fig. 1–3. (a) and (b) show S 2p spectra taken at two different emission angles of photoelectrons (0° and 60° normal to the surface for (a) and (b), respectively). | ||
For comparison, oxidation of a P3HT layer on a bare ITO substrate was also evaluated (Fig. 5). The comparison of the oxidation behaviors of P3HT on ALD-ZnO/ITO and bare ITO in Fig. 4a and 5 clearly reveals that the oxidation behavior of the organic polymer is influenced by the substrate where P3HT is much less oxidized on bare ITO than on ALD-ZnO/ITO. This result is summarized in Table 3.
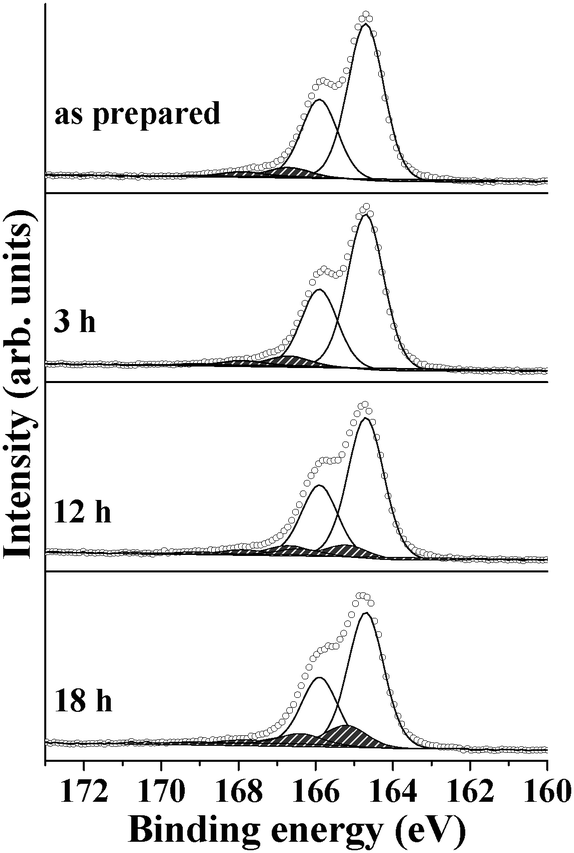 | ||
| Fig. 5 P3HT oxidation on bare ITO was studied under the same conditions as the samples shown in Fig. 4. | ||
| Non-oxidizeda (164.7 eV) | Sulfoxidea (165.2 eV) | Sulfonea (166.7 eV) | Total-oxidizeda (∼170.0 eV) | ||
|---|---|---|---|---|---|
| Unit: %.a Binding energy of the 2p3/2 state of S. | |||||
| ALD-ZnO | As prepared | 93.6 | 0.0 | 6.4 | 0.0 |
| 3 h | 90.7 | 3.5 | 5.4 | 0.4 | |
| 12 h | 82.9 | 11.7 | 4.8 | 0.6 | |
| 18 h | 69.4 | 24.6 | 4.5 | 1.5 | |
| Bare ITO | As prepared | 93.2 | 0.0 | 6.2 | 0.6 |
| 3 h | 92.5 | 0.0 | 6.5 | 1.0 | |
| 12 h | 84.4 | 8.1 | 6.0 | 1.5 | |
| 18 h | 79.5 | 13.9 | 5.1 | 1.5 | |
It is notable that the oxidation of P3HT preferentially takes place at the topmost layers of P3HT, which is far away from the P3HT/oxide interfaces. However, the degree of oxidation is governed by the structure of the substrate. The oxidation behaviors of P3HT on bare and TiO2-covered ZnO ripples are much different and those on bare and ZnO-deposited ITO with flat surface structures are also dissimilar. It is important to mention that the electronic and optical properties of ZnO ripple structures have been suggested to be strongly altered by additional ALD-TiO2 layers where intrinsic defect sites of ZnO ripple structures can act as the recombination centers of optically created electron–hole pairs and TiO2 layers can heal the defect sites, reducing electron–hole recombination at the ZnO surfaces.24,28 One can suggest the following mechanism for the oxidation of P3HT on ZnO ripples: light absorbed by the P3HT layer creates electron–hole pairs in P3HT, and these pairs can interact with O2 and H2O to form O2− and OH radicals, which oxidize the P3HT layer.20–22 When a highly defective ZnO surface exists between ZnO and P3HT, electron–hole pairs created in P3HT can diffuse into the interface and recombination of the electrons and holes takes place before O2− and OH radicals are formed. When TiO2 layers are formed between ZnO and P3HT, the electron–hole pairs created in P3HT can survive with a longer lifetime since recombination centers on ZnO are healed by TiO2. This can then increase the probability that O2 and H2O interact with electrons and holes to form strong oxidizing agents. This picture can be also valid for bare and flat ZnO-deposited ITO since electron–hole pairs created in P3HT by light absorption can be much more efficiently recombined at metallic ITO, whereas semi-conductive ZnO tends to selectively accept electrons and retard holes, reducing the electron–hole recombination rate.
4. Conclusion
In this study, we demonstrated that P3HT on ZnO oxidizes under humid air and visible light conditions mostly into sulfoxide and C–OH species with a sustained ring structure. The ALD-deposited TiO2 layer between ZnO and P3HT significantly increases the degree of oxidation of P3HT under the same conditions. It is expected that P3HT oxidation could take place at the P3HT/oxide interface since the oxidation behavior of P3HT is significantly changed by altering the structure of the oxide substrate. Using a model system of P3HT on a flat ZnO layer prepared by ALD and angle-resolved photoelectron spectroscopy, it was evidenced that the topmost layers of P3HT, not P3HT buried at P3HT/oxide interfaces, are preferentially oxidized. This result is in contrast to our aforementioned expectation. Our results can be explained considering the lifetimes of electron–hole pairs optically created in P3HT, which can be significantly altered by the structure of the substrate (or P3HT/oxide interfaces).Acknowledgements
This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (2012R1A1B3000992). This research was also supported by a grant from KIMS.References
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lussem and K. Leo, Nature, 2009, 459, 234–238 CrossRef CAS PubMed.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- J. Y. Kim, K. Lee, N. E. Coates, D. Moses, T. Q. Nguyen, M. Dante and A. J. Heeger, Science, 2007, 317, 222–225 CrossRef CAS PubMed.
- C. Cabanetos, A. El Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. J. Frechet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656–4659 CrossRef CAS PubMed.
- M. Jorgensen, K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 686–714 CrossRef PubMed.
- A. Guerrero, P. P. Boix, L. F. Marchesi, T. Ripolles-Sanchis, E. C. Pereira and G. Garcia-Belmonte, Sol. Energy Mater. Sol. Cells, 2012, 100, 185–191 CrossRef CAS PubMed.
- H. Hintz, H. J. Egelhaaf, L. Luer, J. Hauch, H. Peisert and T. Chasse, Chem. Mater., 2011, 23, 145–154 CrossRef CAS.
- U. Aygul, H. Hintz, H. J. Egelhaaf, A. Distler, S. Abb, H. Peisert and T. Chasse, J. Phys. Chem. C, 2013, 117, 4992–4998 Search PubMed.
- H. Hintz, H. J. Egelhaaf, H. Peisert and T. Chasse, Polym. Degrad. Stab., 2010, 95, 818–825 CrossRef CAS PubMed.
- M.-G. Jeong, H. O. Seo, D. H. Kim, K.-D. Kim, E. J. Park, Y. D. Kim and D. C. Lim, J. Phys. Chem. C, 2014, 118, 3483–3489 CAS.
- M. Manceau, A. Rivaton, J. L. Gardette, S. Guillerez and N. Lemaitre, Polym. Degrad. Stab., 2009, 94, 898–907 CrossRef CAS PubMed.
- K. Norrman, M. V. Madsen, S. A. Gevorgyan and F. C. Krebs, J. Am. Chem. Soc., 2010, 132, 16883–16892 CrossRef CAS PubMed.
- M. V. Madsen, T. Tromholt, A. Bottiger, J. W. Andreasen, K. Norrman and F. C. Krebs, Polym. Degrad. Stab., 2012, 97, 2412–2417 CrossRef CAS PubMed.
- M. Manceau, S. Chambon, A. Rivaton, J. L. Gardette, S. Guillerez and N. Lemaitre, Sol. Energy Mater. Sol. Cells, 2010, 94, 1572–1577 CrossRef CAS PubMed.
- S. Chambon, A. Rivaton, J.-L. Gardette, M. Firon and L. Lutsen, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 317–331 CrossRef CAS.
- J. Alstrup, K. Norrman, M. Jørgensen and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2006, 90, 2777–2792 CrossRef CAS PubMed.
- S. K. Hau, H. L. Yip, N. S. Baek, J. Y. Zou, K. O'Malley and A. K. Y. Jen, Appl. Phys. Lett., 2008, 92, 253301 CrossRef PubMed.
- S. Y. Park, H. O. Seo, K.-D. Kim, W. H. Shim, J. Heo, S. Cho, Y. D. Kim, K. H. Lee and D. C. Lim, J. Phys. Chem. C, 2012, 116, 15348–15352 CAS.
- A. M. Peiro, G. Doyle, A. Mills and J. R. Durrant, Adv. Mater., 2005, 17, 2365–2368 CrossRef CAS.
- L. Xiao-e, A. N. M. Green, S. A. Haque, A. Mills and J. R. Durtant, J. Photochem. Photobiol., A, 2004, 162, 253–259 CrossRef PubMed.
- J. M. Coronado, A. Javier Maira, A. Martínez-Arias, J. C. Conesa and J. Soria, J. Photochem. Photobiol., A, 2002, 150, 213–221 CrossRef CAS.
- D. C. Lim, W. H. Shim, K.-D. Kim, H. O. Seo, J. H. Lim, Y. Jeong, Y. D. Kim and K. H. Lee, Sol. Energy Mater. Sol. Cells, 2011, 95, 3036–3040 CrossRef CAS PubMed.
- K.-D. Kim, D. C. Lim, H. O. Seo, J. Y. Lee, B. Y. Seo, D. J. Lee, Y. Song, S. Cho, J. H. Lim and Y. D. Kim, Appl. Surf. Sci., 2013, 279, 380–383 CrossRef CAS PubMed.
- H. O. Seo, M.-G. Jeong, K.-D. Kim, D. H. Kim, Y. D. Kim and D. C. Lim, Surf. Interface Anal., 2014, 46, 544–549 CrossRef CAS.
- M. Manceau, J. Gaume, A. Rivaton, J. L. Gardette, G. Monier and L. Bideux, Thin Solid Films, 2010, 518, 7113–7118 CrossRef CAS PubMed.
- J. Heeg, C. Kramer, M. Wolter, S. Michaelis, W. Plieth and W. J. Fischer, Appl. Surf. Sci., 2001, 180, 36–41 CrossRef CAS.
- K.-D. Kim, D. C. Lim, J. Hu, J. D. Kwon, M.-G. Jeong, H. O. Seo, J. Y. Lee, K. Y. Jang, J. H. Lim and K. H. Lee, et al. , ACS Appl. Mater. Interfaces, 2013, 5, 8718–8723 CAS.
- S. Y. Park, H. O. Seo, K.-D. Kim, J. E. Lee, J. D. Kwon, Y. D. Kim and D. C. Lim, Phys. Status Solidi RRL, 2012, 6, 196–198 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Physical Electronics, Inc., Minesota, 1995 Search PubMed.
- T. Schwieger, X. Liu, H. Peisert, B. Adolphi, N. Kiriy and M. Knupfer, J. Appl. Phys., 2005, 97, 123712 CrossRef PubMed.
- X. T. Hao, T. Hosokai, N. Mitsuo, S. Kera, K. K. Okudaira, K. Mase and N. Ueno, J. Phys. Chem. B, 2007, 111, 10365–10372 CrossRef CAS PubMed.
- G. Ertl and J. Kuppers, Low Energy Electrons and Surface Chemistry, VCH, 1985 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp03665d |
| This journal is © the Owner Societies 2015 |