
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1D iron(II) spin crossover coordination polymers with 3,3′-azopyridine – kinetic trapping effects and spin transition above room temperature†
Sophie
Schönfeld
,
Charles
Lochenie
,
Peter
Thoma
and
Birgit
Weber
*
Inorganic Chemistry II, University of Bayreuth, Universitätsstr. 30, 95440 Bayreuth, Germany. E-mail: weber@uni-bayreuth.de; Tel: +49(0)921 55 2555
First published on 22nd May 2015
Abstract
Two new iron(II) 1D coordination polymers with spin crossover behaviour were synthesised using 3,3′-azopyridine as bridging axial ligand and N2O2 coordinating Schiff base-like equatorial ligands. The X-ray structures of both complexes were solved revealing very different packing patterns for the two complexes. Magnetic measurements reveal a spin transition with hysteresis and kinetic trapping effects at lower temperatures for one complex and a spin transition above room temperature for the second complex.
Introduction
Iron(II) spin crossover (SCO) complexes belong to a very actively investigated class of switchable molecular materials. The transition between the paramagnetic high spin (HS, S = 2) and the diamagnetic low spin (LS, S = 0) state can be triggered by many different possibilities.1 Additionally these complexes can be nanostructured,2 incorporated into composite materials3 or can be combined with additional properties.4 These lead to a very wide application potential for this kind of complexes in the field of sensing or data storage. From the application point of view, abrupt spin transitions (that take place in the range of a few K) or spin transitions with hysteresis (with different transition temperatures T1/2 in the heating and the cooling modes) are especially desirable. For some of these complexes with cooperative spin transition, the scan rate used for the magnetic measurement can influence the observed width of the thermal hysteresis loop. A very pronounced scan rate dependence was recently reported for a dinuclear iron(II) spin crossover complex5 and a mononuclear cobalt(II) complex,6 both showing a spin transition with hysteresis. Often phase transitions are responsible for a pronounced scan rate dependence of the width of the thermal hysteresis loop. A very impressive example, where depending on the scan rate different LS states with different metastable HS states were obtained, was presented by Real and co-workers.7 Another kinetic effect can be observed, if the thermal spin transition takes place at rather low temperatures. In this case the interplay between a kinetically slow thermal spin transition and the relaxation from the metastable HS state is possible.8,9For the spin crossover complexes investigated by our group, the observation of kinetic effects is rare. Routinely, we present the results of the magnetic measurements in settle mode to allow a better comparison of the different substances.10 However, the first characterisation of the magnetic properties of a new compound is normally done in sweep mode (usually with a rate of 5 K min−1) and in most of the cases no significant differences between the two curves are observed. So far there is only one example for a kinetic trapping effect (close T1/2 and TLIESST) observed for a 1D chain complex synthesised by our group.11 Here we present two new 1D chain coordination polymers of the Schiff base-like ligands used by our group and 3,3′-azopyridine as bridging axial ligand. While one of the two complexes shows a spin transition above room temperature, for the second complex pronounced kinetic effects are observed. The differences in the magnetic properties can be related to the differences in the crystal packing of these two complexes.
Results
Synthetic procedures
The octahedral iron(II) 1D coordination polymers were obtained in a one-pot reaction using the related mononuclear methanol complexes as starting materials.12,13 A ligand substitution reaction with the bridging ligand 3,3′-azopyridine14,15 gave complexes {[FeL1(azpy)]·0.5(azpy)}n (1) and {[FeL2(azpy)]}n (2) in good yields. Scheme 1 displays the general synthesis of the iron(II) coordination polymers 1 and 2. The complexes were fully characterised by elemental analysis, IR and mass spectrometry. Moreover X-ray diffraction data could be obtained for both complexes. | ||
| Scheme 1 General synthesis of the iron(II) coordination polymers 1 and 2. | ||
Single crystal X-ray structure analysis
Crystals suitable for X-ray structure analysis were obtained for 1 and 2 by using a slow diffusion technique at room temperature. The crystallographic data of 1 and 2 are summarized in Table S1.† The molecular structures of both compounds were determined at 133 K.Complex 1 crystallises in monoclinic space group C2/c, with eight formula units in the unit cell. Fig. 1 displays the ORTEP drawing of the asymmetric unit together with the used atom numbering scheme. The asymmetric unit of 1 consists of one monomeric unit of the coordination polymer {[FeL1(azpy)]}n and half of a non-coordinating azpy molecule. This is in agreement with the composition obtained for the bulk material (see the Experimental section). The iron(II) centre has an octahedral coordination sphere consisting of the equatorially coordinated Schiff base-like ligand and the axially coordinated bridging ligand azpy, bound through terminal 3-pyridyl groups. Selected bond lengths and angles within the inner coordination sphere of both complexes 1 and 2 are summarized in Table 1. The bond lengths and angles within the inner coordination sphere of 1 are within the range reported for HS iron(II) complexes of this ligand type.11,16,17 The average bond lengths are 2.02 Å (Fe–Oeq), 2.10 Å (Fe–Neq) and 2.26 Å (Fe–Nax). The average Oeq–Fe–Oeq angle is 110.58(9)° clearly in the range typical for the HS state.11,16,17 The Nax–Fe–Nax angle (173.18(10)°) does not differ significantly from the expected 180° for an ideal octahedron.
 | ||
| Fig. 1 ORTEP drawing of the asymmetric unit of 1 with the atom numbering scheme used in the text. Hydrogen atoms are omitted for clarity. Thermal ellipsoids presented at 50% level. | ||
| Fe–Neq | Fe–Oeq | Fe–Nax | Oeq–Fe–Oeq | Nax–Fe–Nax | |
|---|---|---|---|---|---|
| 1 | 2.098(3) | 2.019(2) | 2.257(3) | 110.58(9) | 173.18(10) |
| 2.109(3) | 2.024(2) | 2.266(3) | |||
| 2 | 1.903(2) | 1.9341(19) | 1.997(2) | 88.74(7) | 174.91(9) |
| 1.893(2) | 1.9322(19) | ||||
Complex 2 crystallizes in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with two formula units in the unit cell. Fig. 2 displays the ORTEP drawing of the complex together with the atomic numbering scheme. The asymmetric unit consists of one monomeric unit of the coordination polymer {[FeL2(azpy)]}n. The bond lengths and angles of the inner coordination sphere of 2, displayed in Table 1, are within the range reported for a octahedral LS iron(II) complex of this ligand type.11,16,17 The average bond lengths are 1.90 Å (Fe–Oeq), 1.93 Å (Fe–Neq) and 2.00 Å (Fe–Nax). Based on the Oeq–Fe–Oeq angle it is possible to make a statement about the spin state of the iron centre. For this complex the average angle is 89° clearly in the range typical for the LS state.11,16,17 The Nax–Fe–Nax angle (174.91(9)°) does not differ significantly from the expected 180° for an ideal octahedron.
, with two formula units in the unit cell. Fig. 2 displays the ORTEP drawing of the complex together with the atomic numbering scheme. The asymmetric unit consists of one monomeric unit of the coordination polymer {[FeL2(azpy)]}n. The bond lengths and angles of the inner coordination sphere of 2, displayed in Table 1, are within the range reported for a octahedral LS iron(II) complex of this ligand type.11,16,17 The average bond lengths are 1.90 Å (Fe–Oeq), 1.93 Å (Fe–Neq) and 2.00 Å (Fe–Nax). Based on the Oeq–Fe–Oeq angle it is possible to make a statement about the spin state of the iron centre. For this complex the average angle is 89° clearly in the range typical for the LS state.11,16,17 The Nax–Fe–Nax angle (174.91(9)°) does not differ significantly from the expected 180° for an ideal octahedron.
 | ||
| Fig. 2 ORTEP drawing of the asymmetric unit of 2 with the atom numbering scheme used in the text. Hydrogen atoms are omitted for clarity. Thermal ellipsoids presented at 50% level. | ||
Table 2 gives the selected bond lengths between the carbon atoms of the Schiff base-like ligand for both compounds. The average bond length of 1.43 Å cannot be attributed to a single or double bond, showing that the negative charge of the nitrogen is therefore delocalized over the chelate ring.
The comparison of the torsion angles (Table 2) between the coordinating azpy molecule and the non-coordinating azpy of 1 reveals that the coordinating molecule is bent whereas the non-coordinating one is ideally flat as expected for a sp2 hybrid system. By connecting the two iron(II) centers the molecule is slightly bent and therefore the torsion angles differ from the expected 180°. However, this impact is not as pronounced for the bridging azpy molecules of 2. Here the torsion angle corresponds to that of an ideally flat molecule and there appears to be less strain in the system.
In Fig. 3, the packing of the chains in the crystal of compound 1 is displayed. The 1D chains of 1 exhibit a zigzag motif. Chains that propagate in the same direction are linked by a hydrogen bond between the coordinating and the non-coordinating azpy (C55–H55⋯N5). The relevant intermolecular distances are summarized in Table 3. The 1D chains in combination with the hydrogen bonds form a 2D plane. The planes themselves are again linked by hydrogen bonds, on the one hand directly between the complexes (C8–H8⋯O3, C25–H25⋯O4) and on the other hand through the non-coordinating azpy (C52–H52⋯O4). These specific linkages are responsible for the unique perpendicular orientation of the chains. Interestingly, such a perpendicular arrangement of the 1D chains was also observed for related 4,4′-azopyridine-based coordination polymers,18,19 whereas for other coordination polymers of this ligand type, only parallel aligned 1D chains were obtained so far.16
 | ||
| Fig. 3 Molecular packing of compound 1 in the crystal at 133 K. Top: view along [−1 1 0] and bottom: view along [1 1 0]. The hydrogen atoms have been omitted for clarity. | ||
| Donor–H⋯acceptor | D–H | H⋯A | D⋯A | D–H⋯A | |
|---|---|---|---|---|---|
| a x, 2 − y, 1/2 + z. b x, 2 − y, −1/2 + z. c 3/2 − x, −1/2 + y, 3/2 − z. d 2 − x, 1 − y, 1 − z. | |||||
| 1 | C(8)–H(8)⋯O(3)a | 0.93 | 2.57 | 3.406(4) | 150 |
| C(25)–H(25)⋯O(4)b | 0.93 | 2.44 | 3.221(4) | 141 | |
| C(52)–H(52)⋯O(4) | 0.93 | 2.40 | 3.280(6) | 158 | |
| C(55)–H(55)⋯N(5)c | 0.93 | 2.51 | 3.440(6) | 173 | |
| 2 | C(27)–H(27)⋯O(5)d | 0.95 | 2.56 | 3.261(3) | 131 |
In contrast to 1, in the crystal packing of 2, the 1D chains propagate parallel along the [1 0 0] direction and are stacked such that the unit cell contains no residual solvent-accessible void volume (Fig. 4). The parallel 1D chains are linked by a hydrogen bond between the coordinating azpy and the oxygen of the ester group of the equatorial ligand (C27–H27⋯O5, see Table 3). The 1D chain of 2 exhibits also a slight zigzag motif due to the trans-arrangement of the coordinating nitrogen atoms in the ligand.
 | ||
| Fig. 4 Molecular packing of compound 2 in the crystal at 133 K. View along [0 0 1] (top) and [1 0 0] (bottom). The hydrogen atoms have been omitted for clarity. | ||
Magnetic measurements
Magnetic susceptibility measurements in the temperature range from 300/400 K to 10 K were undertaken to follow the iron(II) spin state change for the two samples.The thermal dependence of the χMT product for complex 1 with the “standard” scanning rate of 5 K min−1 showed indications of a kinetic trapping of the HS state. Therefore the compound was studied in sweep mode with different cooling rates (0.5, 2, 5, and 10 K min−1). The results of these measurements are displayed in Fig. 5. At room temperature, the compound has a χMT product of 3.21 cm3 K mol−1, in the typical range for iron(II) complexes in the HS state. Upon cooling with a rate of 0.5 K min−1, the χMT product remains constant until 90 K, where the compound undergoes an incomplete gradual spin crossover from HS to LS to attain a minimum χMT value of 1.78 cm3 K mol−1, indicative of a mixed HS(0.5)/LS(0.5) state. Such a plateau (remaining HS fraction) is often observed for coordination polymers of this ligand type with either a pronounced zigzag structure or a perpendicular arrangement of the 1D chains.11,18,20
 | ||
| Fig. 5 Plot of the χMT product vs. T over the range 150–20 K with 10 K min−1 (squares), 5 K min−1 (filled circles), 2 K min−1 (open circles), 0.5 K min−1 (triangles) rates of cooling for the component 1. | ||
The χMT product stays constant until 10 K. Upon heating, compound 1 exhibits a 15 K wide thermal hysteresis loop with critical temperatures of T1/2↓ = 90 K upon cooling and T1/2↑ = 105 K upon heating, coming back to a full HS state. By cooling down the component with a rate of 2 K min−1 a minimum χMT value of 2.37 cm3 K mol−1 at 100 K can be observed, which is no longer in a range typical for a full HS iron(II) compound, nor for a mixed HS/LS state (74% of active sites remain trapped in the HS state). By increasing the cooling rate to 5 K min−1, a minimum value of 3.06 cm3 K mol−1 at 101 K is detected, showing that 95% of the active sites are trapped in the HS state. Finally, with a cooling rate of 10 K min−1, the χMT values are constant within the whole temperature range and maintain a value of 3.21 cm3 K mol−1, showing that the HS state of the compound 1 is completely trapped.
Since complex 1 shows extraordinary thermal kinetic trapping, a TIESST (temperature induced excited spin state trapping) experiment was conducted (Fig. 6). The compound was rapidly cooled with a rate of 10 K min−1 from 300 K to 10 K. Using this procedure, the complex got trapped in a metastable HS state at 10 K. At this temperature the system would be in a mixed HS/LS form if a very slow cooling rate was used. The temperature is then slowly increased at a rate of 0.5 K min−1 and the magnetic behaviour is recorded. The magnetic response of the trapped high spin state remains relatively constant up to 60 K. Above 60 K the system reaches the thermally activated domain, and the HS → LS relaxation becomes more and more rapid to reach a minimum in the χMT vs. T curve at 85 K with a χMT value of 1.78 cm3 K mol−1, corresponding to the mixed HS/LS state. The critical temperature TTIESST was found to be 73 K (the inset in Fig. 6). This is significantly higher than the TLIESST temperatures observed so far for complexes of this ligand type (assuming that these temperatures are usually in the same order of magnitude). Upon further heating, the compound goes back to a full HS state at 105 K. In order to determine the actual width of the thermal hysteresis loop of 1 without any kinetic trapping effects, the complex was cooled from room temperature to a temperature between 80 K and 105 K (in intervals of 5 K) and then left at this temperature for the HS → LS relaxation (open triangles in Fig. 6). To follow the relaxation process, every 10 min a measurement point was taken. As illustrated in Fig. 6, full relaxation is observed up to a temperature of 95 K. At 100 K the system no longer relaxes completely into the mixed HS/LS state and at 105 K the complex remains in the HS state. This experiment shows that even with a sweeping velocity of 0.5 K min−1 kinetic effects influence the width of the hysteresis loop (determined to be 15 K) and the actual width excluding kinetic effects is more in the region of 7 K.
 | ||
| Fig. 6 Black cycles: TIESST experiment recorded for the coordination polymer 1 at a warming rate 0.5 K min−1. The inset shows the first deviation for the determination of TTIESST. Open triangles: relaxation of the thermally trapped HS state at 80, 85, 90, 95, 100 and 105 K for the determination of the hysteresis width without of trapping effects. The solid lines are guides for the eye. | ||
The thermal dependence of the χMT product of 2 is displayed in Fig. 7. The complex shows a gradual SCO between 350 and 400 K with a transition temperature of T1/2 = 355 K to reach a maximum χMT value of 3.11 cm3 K mol−1, typical for an iron(II) complex in the HS state. Between 340 and 10 K the χMT values are approximately constant with a value close to zero (0.01 cm3 K mol−1 at 10 K), which is typical for an iron(II) complex in the LS state.
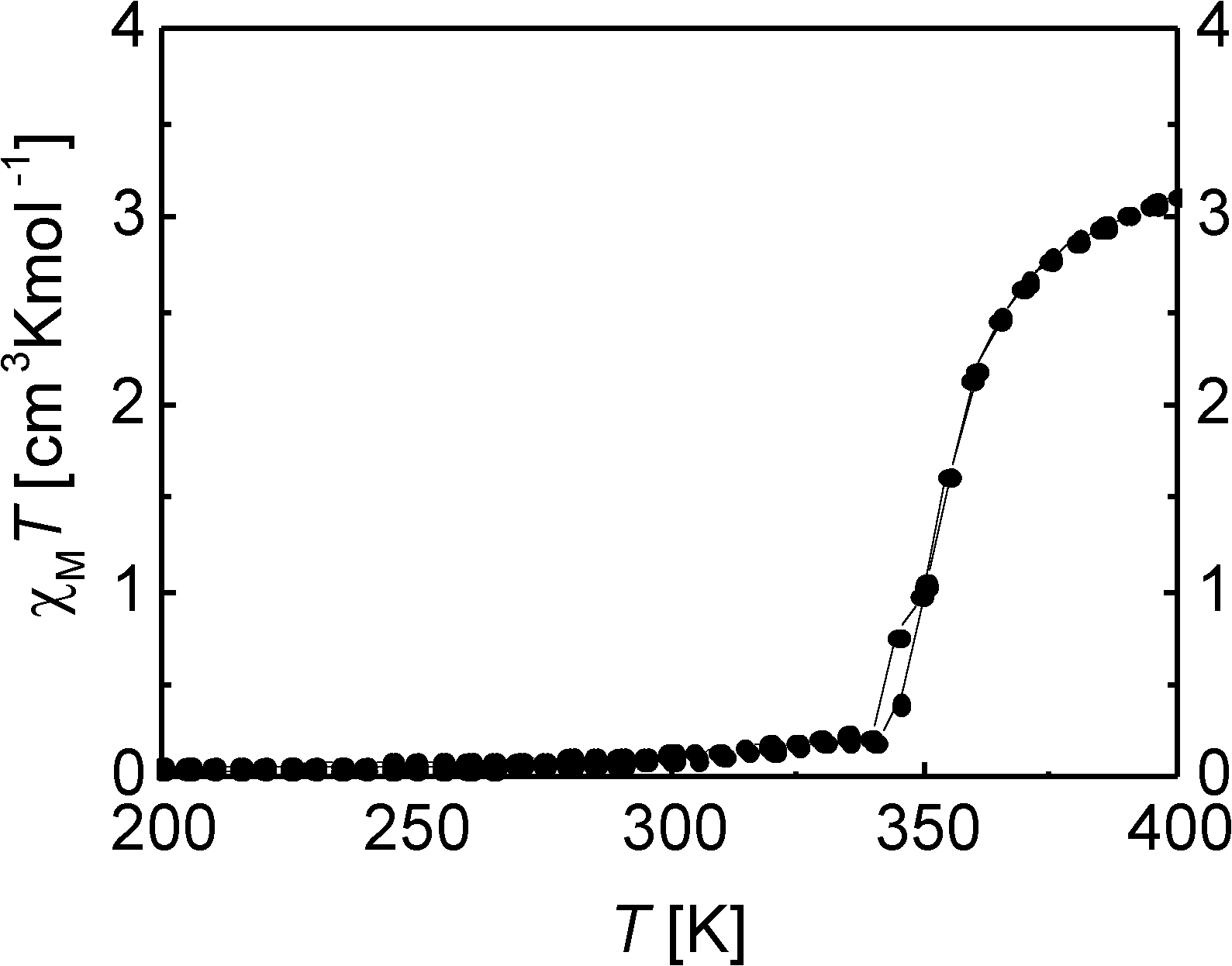 | ||
| Fig. 7 Plot of the χMT product vs. T over the range 200–400 K for the compound 2. | ||
Discussion
The two complexes 1 and 2 vary only in one substituent that has a marginal influence on the overall ligand field strength at the iron centre. Investigations into the spin transition behaviour in solution revealed almost identical transition temperatures.16 Despite this, the spin transition behaviour, considering the transition temperature T1/2, the completeness of the spin transition and the amount of cooperative interactions (hysteresis or not), of the two complexes is very different. This can only be due to the very different packing of the 1D chains in the crystal. The difference in the cooperative interactions (7 K wide hysteresis for 1vs. gradual spin crossover for 2) is probably the easiest to be explained. In contrast to 2 the packing of the molecules of 1 in the crystal (Fig. 3) reveals four different hydrogen bonds leading to 2D layers that are further interconnected. The hydrogen bonding of the free azpy molecules plays a decisive role. The additional azpy molecules in the packing of the complex cross-link these perpendicularly oriented chains that propagate in the same direction with a strong hydrogen bond (Table 3, hydrogen bond 4). In contrast to this, in the case of 2 only one non-classical hydrogen bond is observed. This significant difference in the amount of hydrogen bonds between the 1D chains easily explains the difference in the cooperative interactions. The magnetic characterisation for complex 1 shows an incomplete spin transition that stops at a plateau with about 50% of the molecules in the HS and 50% in the LS states while the SCO of 2 is complete. Two different reasons can be responsible for this observation. The first possibility is that only one out of the two planes propagating in perpendicular directions is undergoing the spin crossover. The second alternative, which was already observed for similar coordination polymers of this ligand type, is the formation of a mixed HS–LS chain with only every second iron centre undergoing spin transition due to restraining steric effects.11 Both possibilities can explain the magnetic behaviour, resulting in a mixed HS/LS state. For 1, intermolecular restraining interactions are easily imaginable because of the zigzag motif of the 1D chains in combination with the perpendicular orientation and the hydrogen bonds. All complexes characterised so far with such a perpendicular arrangement of 1D chains showed steps in the transition curve.18 To provide assurance with regard to this hypothesis, it would be necessary to examine the X-ray diffraction data at low temperatures where the complex is in the mixed HS/LS state. Unfortunately the spin transition takes place at such low temperatures that it was not possible to obtain the desired data. The very low transition temperature of 1, in combination with the intermolecular restraining interactions, also explains the trapping of the HS state during rapid cooling and the decreasing hysteresis width with slow cooling rates. Most likely the spin transition is accompanied by phase transitions (e.g. rearrangement of the H-bond network) leading to these kinetic effects. The complete spin transition of 2 and the weak cooperative effects are in good agreement in the presence of only one hydrogen bond linking the 1D chains that are, additionally, all parallel. Due to the high transition temperature (above room temperature) no kinetic trapping effects are observed.Experimental
Materials and methods
All syntheses were carried out under argon using Schlenk tube techniques. Methanol was dried over magnesium and distilled under argon before use. The syntheses of the starting complexes [FeL1/2(MeOH)2] were prepared as described in the literature.12,13 3,3′-Azopyridine as an axial ligand was prepared according to the literature.14,15![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1638(s, ν[CO]), 1552(s, ν[N
= 1638(s, ν[CO]), 1552(s, ν[N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N]); MS (DEI-(+), 70 eV): m/z (%): 382 (100) [FeL1+], 367 (47) [M+–Me], 184 (76) [azpy+], 106 (46) [C5H4N3+], 78 (100) [C5H4N+], 51 (37) [C4H3+]. Elemental analysis calcd (%) for C33H30FeN8O4 (658.50 g mol−1) C 60.19, H 4.59, N 17.02; found: C 60.52, H 4.48, N 17.07.
= 1688 (s, ν[CO]), 1557 (s, ν[N
N]); MS (DEI-(+), 70 eV): m/z (%): 382 (100) [FeL1+], 367 (47) [M+–Me], 184 (76) [azpy+], 106 (46) [C5H4N3+], 78 (100) [C5H4N+], 51 (37) [C4H3+]. Elemental analysis calcd (%) for C33H30FeN8O4 (658.50 g mol−1) C 60.19, H 4.59, N 17.02; found: C 60.52, H 4.48, N 17.07.
= 1688 (s, ν[CO]), 1557 (s, ν[N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N]), 1267 (vs, ν[C–O]); MS (DEI-(+), 70 eV): m/z (%): 414 (100) [FeL2+], 382 (29) [M+–OCH3], 340 (46) [M+–COOMe2], 309 (40) [C14H9FeN2O3+], 184 (71) [azpy+], 106 (46) [C5H4N3+], 78 (100) [C5H4N+], 51 (39) [C4H3+]. Elemental analysis calcd (%) for C28H26FeN6O6 (598.39 g mol−1) C 56.20, H 4.38, N 14.04; found: 56.12, H 4.04, N 13.98.
N]), 1267 (vs, ν[C–O]); MS (DEI-(+), 70 eV): m/z (%): 414 (100) [FeL2+], 382 (29) [M+–OCH3], 340 (46) [M+–COOMe2], 309 (40) [C14H9FeN2O3+], 184 (71) [azpy+], 106 (46) [C5H4N3+], 78 (100) [C5H4N+], 51 (39) [C4H3+]. Elemental analysis calcd (%) for C28H26FeN6O6 (598.39 g mol−1) C 56.20, H 4.38, N 14.04; found: 56.12, H 4.04, N 13.98.
X-ray diffraction
The intensity data of 1 and 2 were collected using a Stoe IPDS II diffractometer with graphite-monochromated MoKα radiation. The data were corrected for Lorentz and polarization effects. The structures were solved by direct methods using SIR97 (ref. 21) and refined by full-matrix least-square techniques against F02 (SHELXL-97 (ref. 22)). The hydrogen atoms were included at calculated positions with fixed displacement parameters, and allowed to ride their parent atoms. All non-hydrogen atoms were refined anisotropically. ORTEP-III23 was used for the structure representation and SCHAKAL-99 (ref. 24) for the representation of the molecular packing. CCDC 1053320 (1) and 1053319 (2) contain the supplementary crystallographic data for this paper and can be obtained free of charge from the Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request/cif.Magnetic measurements
Magnetic measurements on the bulk materials were carried out using a SQUID MPMS-XL5 from Quantum Design with an applied field of 5000 G (1), 2000 G (1-TIESST and 2), respectively, and in the temperature range from 400 to 10 K in the sweep (1) and settle (2) modes. The sample was prepared in a gelatine capsule held in a plastic straw. The raw data were corrected for the diamagnetic part of the sample holder and the diamagnetism of the organic ligand using tabulated Pascal's constants.Conclusions
In this work we have presented two novel 1D iron(II) spin crossover coordination polymers with 3,3′-azopyridine as an axial ligand. The results from X-ray structure analysis revealed that the differences in the SCO behavior of the two complexes can be attributed to differences in the formation of hydrogen bonds in the molecular packing. The presence of free ligand molecules in the crystal packing of 1 supports the special perpendicular orientation of the polymer chains which again is associated with the unique magnetic properties. In contrast to the studies so far, where either the hysteresis width5,6 or the completeness of the spin transition (quenching effects)8,9 is influenced by the scan rate, we observe for complex 1 a combination of both effects.Acknowledgements
Financial support from the German Science Foundation (SFB840; project A10) and the University of Bayreuth is acknowledged.Notes and references
- (a) Spin-Crossover Materials, ed. M. A. Halcrow, John Wiley & Sons Ltd, Chichester, 2013 Search PubMed; (b) Spin Crossover in Transition Metal Compounds I–III, ed. P. Gütlich and H. Goodwin, Springer Berlin, Heidelberg, 2004, pp. 233–235 Search PubMed.
- (a) A. Tissot, New J. Chem., 2014, 38, 1840 RSC; (b) E. Coronado, J. R. Galán-Mascarós, M. Monrabal-Capilla, J. García-Martínez and P. Pardo-Ibáñez, Adv. Mater., 2007, 19, 1359–1361 CrossRef CAS PubMed; (c) F. Volatron, L. Catala, E. Rivière, A. Gloter, O. Stéphan and T. Mallah, Inorg. Chem., 2008, 47, 6584–6586 CrossRef CAS PubMed; (d) I. Boldog, A. B. Gaspar, V. Martínez, P. Pardo-Ibañez, V. Ksenofontov, A. Bhattacharjee, P. Gütlich and J. A. Real, Angew. Chem., 2008, 120, 6533–6537 CrossRef PubMed.
- (a) M. Cavallini, I. Bergenti, S. Milita, G. Ruani, I. Salitros, Z.-R. Qu, R. Chandrasekar and M. Ruben, Angew. Chem., 2008, 120, 8724–8728 CrossRef PubMed; (b) C. Thibault, G. Molnár, L. Salmon, A. Bousseksou and C. Vieu, Langmuir, 2010, 26, 1557–1560 CrossRef CAS PubMed; (c) A. D. Naik, L. Stappers, J. Snauwaert, J. Fransaer and Y. Garcia, Small, 2010, 6, 2842–2846 CrossRef CAS PubMed; (d) Y. Raza, F. Volatron, S. Moldovan, O. Ersen, V. Huc, C. Martini, F. Brisset, A. Gloter, O. Stephan, A. Bousseksou, L. Catala and T. Mallah, Chem. Commun., 2011, 47, 11501–11503 RSC; (e) C. Göbel, T. Palamarciuc, C. Lochenieand and B. Weber, Chem. – Asian J., 2014, 9, 2232–2238 CrossRef PubMed.
- (a) S. Titos-Padilla, J. M. Herrera, X.-W. Chen, J. J. Delgado and E. Colacio, Angew. Chem., Int. Ed., 2011, 50, 3290–3293 CrossRef CAS PubMed; (b) L. Salmon, G. Molnár, D. Zitouni, C. Quintero, C. Bergaud, J.-C. Micheau and A. Bousseksou, J. Mater. Chem., 2010, 20, 5499 RSC; (c) A. B. Gaspar and M. Seredyuk, Coord. Chem. Rev., 2014, 268, 41–58 CrossRef CAS PubMed.
- R. Kulmaczewski, J. Olguín, J. A. Kitchen, H. L. C. Feltham, G. N. L. Jameson, J. L. Tallon and S. Brooker, J. Am. Chem. Soc., 2014, 136, 878–881 CrossRef CAS PubMed.
- R. G. Miller, S. Narayanaswamy, J. L. Tallon and S. Brooker, New J. Chem., 2014, 38, 1932 RSC.
- M. Seredyuk, M. C. Muñoz, M. Castro, T. Romero-Morcillo, A. B. Gaspar and J. A. Real, Chem. – Eur. J., 2013, 19, 6591–6596 CrossRef CAS PubMed.
- V. A. Money, C. Carbonera, J. Elhaïk, M. A. Halcrow, J. A. K. Howard and J.-F. Létard, Chem. – Eur. J., 2007, 13, 5503–5514 CrossRef CAS PubMed.
- N. F. Sciortino, S. M. Neville, C. Desplanches, J.-F. Létard, V. Martinez, J. A. Real, B. Moubaraki, K. S. Murray and C. J. Kepert, Chem. – Eur. J., 2014, 20, 7448–7457 CrossRef CAS PubMed.
- (a) W. Bauer, C. Lochenieand and B. Weber, Dalton Trans., 2014, 43, 1990–1999 RSC; (b) C. Lochenie, W. Bauer, A. P. Railliet, S. Schlamp, Y. Garcia and B. Weber, Inorg. Chem., 2014, 53, 11563–11572 CrossRef CAS PubMed; (c) R. Nowak, W. Bauer, T. Ossianderand and B. Weber, Eur. J. Inorg. Chem., 2013, 975–983 CrossRef CAS PubMed.
- (a) W. Bauer, W. Scherer, S. Altmannshoferand and B. Weber, Eur. J. Inorg. Chem., 2011, 2803–2818 CrossRef CAS PubMed; (b) C. Baldé, W. Bauer, E. Kaps, C. Desplanches, G. Chastanet, B. Weber and J.-F. Létard, Eur. J. Inorg. Chem., 2013, 2744–2750 CrossRef PubMed.
- W. Bauer, T. Ossiander and B. Weber, Z. Naturforsch., B: J. Chem. Sci., 2010, 323–328 CAS.
- E.-G. Jäger, E. Häussler, M. Rudolph and A. Schneider, Z. Anorg. Allg. Chem., 1985, 525, 67–85 CrossRef PubMed.
- S. Thies, H. Sell, C. Schütt, C. Bornholdt, C. Näther, F. Tuczekand and R. Herges, J. Am. Chem. Soc., 2011, 133, 16243–16250 CrossRef CAS PubMed.
- G. Spaleniak, Z. Daszkiewicz and J. Kyzioł, Chem. Pap., 2009, 63, 313–322 CAS.
- (a) B. Weber, Coord. Chem. Rev., 2009, 253, 2432–2449 CrossRef CAS PubMed; (b) B. Weber and F.-A. Walker, Inorg. Chem., 2007, 46, 6794–6803 CrossRef CAS PubMed.
- S. Schlamp, P. Thoma and B. Weber, Chem. – Eur. J., 2014, 20, 6462–6473 CrossRef CAS PubMed.
- W. Bauer, T. Pfaffeneder, K. Achterhold and B. Weber, Eur. J. Inorg. Chem., 2011, 3183–3192 CrossRef CAS PubMed.
- B. Weber, W. Bauer, T. Pfaffeneder, M. M. Dîrtu, A. D. Naik, A. Rotaruand and Y. Garcia, Eur. J. Inorg. Chem., 2011, 3193–3206 CrossRef CAS PubMed.
- T. M. Pfaffeneder, S. Thallmair, W. Bauer and B. Weber, New J. Chem., 2011, 35, 691–700 RSC.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidoriand and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- (a) C. K. Johnson and M. N. Burnett, ORTEP-III, Oak-Ridge National Laboratory, Oak-Ridge, TN, 1996 Search PubMed; (b) L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- E. Keller, Schakal-99, University of Freiburg, Freiburg, Germany, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 10533191053320. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce00800j |
| This journal is © The Royal Society of Chemistry 2015 |