
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper pyromellitates: a complex story†‡
William
Clegg
*,
James M.
Holcroft§
and
Nicola C.
Martin¶
School of Chemistry, Newcastle University, Newcastle upon Tyne NE1 7RU, UK. E-mail: bill.clegg@ncl.ac.uk
First published on 9th March 2015
Abstract
Reaction of copper(II) salts with pyromellitic acid (benzene-1,2,4,5-tetracarboxylic acid) and bases using slow vapour and gel diffusion techniques produces a variety of complexes containing copper and pyromellitate ions and solvent molecules, both coordinated (usually as terminal ligands) and uncoordinated. In some cases molecules of base are also coordinated, and organic or alkali metal cations may be present. Most of the complexes are 2- or 3-dimensional polymeric networks as expected, though there are also some discrete molecular complex ions, which are unusual for pyromellitate complexes of metals. Coordination of copper(II) is most often square pyramidal with a long apical bond, but with some examples also of square-planar coordination supplemented by additional secondary interactions with carboxylates or solvent molecules above and below the plane to give distorted octahedral geometry. Pyromellitate is fully deprotonated in all but one product, and usually coordinates to 2, 4 or 6 copper ions, and additionally to 8 alkali metal ions where these are present. Hydrogen bonding plays a significant role in all the structures, its role ranging from formation of discrete clusters of coordinated and uncoordinated water molecules to extensive 3-dimensional networks. The impressive structural variety arises from the combination of rigidity of the benzene ring and individual carboxylate groups with torsional flexibility in the connections of these groups, together with the availability of different coordination geometries for copper(II). The results illustrate the considerable structural versatility of pyromellitate ligands and the unreliability of structural design in benzenepolycarboxylate coordination chemistry.
Introduction
In the early 1980s I (Bill Clegg) began collaborative research with Ken Wade, whom I had first met while I was working on a fixed-term contract in Newcastle and he was in nearby Durham. At first the collaboration was at a distance, but it developed further when I returned to Newcastle from Göttingen in 1984 and continued then for almost a decade, during which there were no crystallographic facilities in the Chemistry Department at Durham. First results were in lithium chemistry1 and led to a long-term fruitful interaction with research groups in Strathclyde (principally Ron Snaith, then Rab Mulvey and Eva Hevia), initiated and encouraged by Ken. Many other applications followed in main-group chemistry and in carborane research, including a particularly attractive cyclic tris-carborane compound that featured on the cover of my undergraduate crystallography textbook2 and was dubbed by Ken a ‘Wagnerian Ring Cycle’, although ‘cluster cycle’ was the nearest to this that could appear in the published report.3A Durham project investigating some mixed copper-zinc carboxylates as potential precursors to catalysts for methanol synthesis, as well as for the structural and magnetic properties of metal benzenepolycarboxylate complexes, provided a few samples for crystallographic study, the results of which have not yet been published except in the PhD thesis of Gavin Walker;4 this work was led by Ken's group, including contributions of Hugh MacBride and Stan Hauptman, who prepared and supplied the samples of copper and mixed copper-zinc pyromellitates for structure determination. At this stage the targeted study of benzenepolycarboxylates as linkers in the formation of metal–organic frameworks and reticular families of porous structures had not yet begun.5 The fascination and beauty of the results of our early Durham-Newcastle collaboration in this area led to a more systematic investigation of transition metal complexes of benzenepolycarboxylic acids in my research group over the following years. The area proved to be very complex, both in terms of the variety of products formed from reactions under different or even very similar or supposedly identical conditions and with respect to the structural properties and relationships of these products, some of them made more challenging by the incidence of structural disorder, twinning and pseudo-symmetry.
We have previously reported some of the results, concerning selected complexes of isophthalic (benzene-1,3-dicarboxylic)6 and mellitic (benzene-1,2,3,4,5,6-hexacarboxylic)7 acids. Here we collect together most of the results we have obtained on copper complexes of pyromellitic (benzene-1,2,4,5-tetracarboxylic) acid, including the original structures obtained in the collaboration with Ken Wade, as an appropriate tribute to his memory and our shared research interests (ESI†).
Pyromellitic acid (H4pm) has been widely used in synthetic and structural research. It is the most symmetrical of the three benzenetetracarboxylic acids. In terms of known crystal structures, it vastly surpasses the other two in entries in the Cambridge Structural Database (CSD).8 In our survey of the CSD using the version with updates to February 2014,7 we found 840 entries for complexes, salts and derivatives of H4pm, this figure being surpassed among the benzenepolycarboxylic acids only by the most symmetrical tricarboxylic acid (trimesic, 1,3,5-substituted), by the three dicarboxylic acids, and by benzoic acid. A major point of structural interest in considering the most symmetrically substituted isomers of increasingly substituted acids in this series is that, in contrast to the lower members terephthalic (1,4) and trimesic (1,3,5) acids, the carboxylate groups in H4pm are prevented from achieving coplanarity with the benzene ring by mutual steric interaction, a factor taken to its extreme in the fully substituted and much less studied mellitic acid. The only exception is the possibility of hydrogen bonding between adjacent carboxylate/carboxylic acid groups and this, leading to coplanarity, is found in some, but by no means all, of the 100 or so structures containing doubly-deprotonated H2pm2− anions.
The CSD (version 5.35 with updates to May 2014) has 136 entries containing copper and partially or fully deprotonated pyromellitate anions (ranging from H3pm− to pm4−; two structures also contain H4pm, but none have the parent acid without anions also present). The structures contain also a wide range of other components, including further ligands coordinated to copper, and many incorporate additional metal ions. For comparison with our own results here, we restrict coverage to those compounds in which no other metal ions are present apart from alkali metals, and in which the other components are solvent molecules and monodentate ligands, leaving aside the structures in which auxiliary ligands serve as bridges between copper centres; there are 28 of these, including a number of reports of the same structure in different publications. The additional ligands are nitrogen heterocycles, ammonia, methylamine, hydroxide, and solvents (water and DMF).
Results and discussion
At the time when the work reported here began, there were only two known crystallographically characterized copper pyromellitate complexes: the two-dimensional polymeric network complex with empirical formula corresponding to a decahydrate Cu2(pm)(OH2)6·4H2O9 and the discrete molecular complex Cu2(pm)(9-ane-N3)2·4H2O, where 9-ane-N3 is the three-fold-symmetric terdentate aza-crown macrocycle 1,4,7-trimethyl-1,4,7-trizacyclononane.10 Many others have appeared since then, and the most relevant are referenced below in the discussion. Some of the more recent reports are of structures that we have ourselves also obtained in this work; in most cases our results represent a significant improvement in precision and interpretation, particularly of hydrogen bonding, and we are able to correct some errors in the previous accounts. One of the complexities we have found in the developing story is that the same product can be obtained by different routes (sometimes during the attempt to prepare a different compound, but with one or more intended reactants finding no place in the resulting structure), while parallel or repeated syntheses using similar or supposedly identical procedures and conditions sometimes lead to two or more different products, these occasionally being obtained as a mixture from a single experiment. Similar observations have been made by others.4,11,12In order to reduce this complexity, and that of the structural aspects, we present our results here in a logical rather than a chronological order, grouping the reported compounds and related literature results under four headings: (a) complexes containing only Cu, pyromellitate and solvent; (b) structures incorporating alkali metal cations; (c) anionic complexes with protonated bases as counterions; (d) complexes in which unprotonated bases serve as ligands. The generation of copper pyromellitate complexes involves the deprotonation of H4pm by an appropriate base so that it can coordinate to the metal. Commonly used bases are ammonia, organic amines (both aliphatic and aromatic), and alkali metal hydroxides or carbonates, and the bases themselves or their cations are incorporated in the structures of groups (b)–(d), whereas they are absent in group (a). In presenting the structures below, the primary graphical representation of each gives the crystallographic asymmetric unit, to which are added sufficient symmetry-equivalent atoms to show the immediate environment of each of the unique metal centres and pyromellitate ligands. Further figures are provided as appropriate, to illustrate key features of the structures. Descriptions include the coordination geometry of copper, the dihedral angles between carboxylate groups and benzene rings of pyromellitate, the coordinating function (usually multiple) of the pyromellitate ligands, the resulting dimensionality of the structure, and the hydrogen bonding. Bond lengths and angles for the copper coordination geometry in all the structures are collected in Table 1. Full details including complete bond lengths and angles, torsion angles, and tables of hydrogen bonding are provided in the ESI.†
| 1 | |||
| Cu–O1 | 2.2319(17) | Cu–O2a | 1.9592(18) |
| Cu–O4b | 1.9589(18) | Cu–O5 | 1.9636(18) |
| Cu–O6 | 1.984(2) | ||
| O1–Cu–O2a | 102.39(7) | O1–Cu–O4b | 88.40(7) |
| O1–Cu–O5 | 91.22(7) | O1–Cu–O6 | 101.58(9) |
| O2a–Cu–O4b | 93.62(8) | O2a–Cu–O5 | 166.19(8) |
| O2a–Cu–O6 | 93.15(8) | O4b–Cu–O5 | 89.02(8) |
| O4b–Cu–O6 | 166.45(9) | O5–Cu–O6 | 81.70(8) |
| Symmetry operations: a = −x, −y + 1, −z; b = x − 1, y − 1, z | |||
| 2 | |||
| Cu–O2 | 1.9553(12) | Cu–O4a | 2.0090(12) |
| Cu–O5 | 1.9548(13) | Cu–O6 | 1.9517(13) |
| Cu–O7 | 2.2432(13) | ||
| O2–Cu–O4a | 175.22(5) | O2–Cu–O5 | 86.44(6) |
| O2–Cu–O6 | 91.15(6) | O2–Cu–O7 | 92.90(5) |
| O4a–Cu–O5 | 88.90(5) | O4a–Cu–O6 | 93.60(6) |
| O4a–Cu–O7 | 86.48(5) | O5–Cu–O6 | 173.57(6) |
| O5–Cu–O7 | 95.36(5) | O6–Cu–O7 | 90.71(5) |
| Symmetry operation: a = −x + 1/2, y − 1/2, −z + 1/2 | |||
| 3 | |||
| Cu–O2 | 1.9555(16) | Cu–O6a | 1.9430(15) |
| Cu–O9 | 2.220(3) | Cu–O10 | 1.986(2) |
| Cu–O11 | 1.985(2) | ||
| O2–Cu–O6a | 176.91(11) | O2–Cu–O9 | 84.38(8) |
| O2–Cu–O10 | 90.82(8) | O2–Cu–O11 | 91.46(9) |
| O6a–Cu–O9 | 92.55(9) | O6a–Cu–O10 | 89.98(8) |
| O6a–Cu–O11 | 88.55(10) | O9–Cu–O10 | 100.16(12) |
| O9–Cu–O11 | 95.19(12) | O10–Cu–O11 | 164.64(10) |
| Symmetry operation: a = x, y − 1, z | |||
| 4 | |||
| Cu–O2 | 1.9354(18) | Cu–O4a | 1.9907(18) |
| Cu–O5b | 1.9771(18) | Cu–O7c | 1.9476(19) |
| O2–Cu–O4a | 91.27(8) | O2–Cu–O5b | 177.13(8) |
| O2–Cu–O7c | 91.03(8) | O4a–Cu–O5b | 86.56(8) |
| O4a–Cu–O7c | 177.28(8) | O5b–Cu–O7c | 91.19(8) |
| Symmetry operations: a = −x + 1, −y, −z + 1; b = x, y, z + 1; c = −x + 1, −y + 1, −z + 1 | |||
| 5 | |||
| Cu–O2 | 1.9397(10) | Cu–O2a | 1.9397(10) |
| Cu–O4b | 1.9551(9) | Cu–O4c | 1.9551(9) |
| O2–Cu–O2a | 84.98(6) | O2–Cu–O4b | 92.64(4) |
| O2a–Cu–O4b | 167.72(4) | O2–Cu–O4c | 167.72(4) |
| O2a–Cu–O4c | 92.64(4) | O4b–Cu–O4c | 92.14(6) |
| Symmetry operations: a = −x + 1, y, −z + 3/2; b = −x + 1, −y + 1, −z + 1; c = x, −y + 1, z + 1/2 | |||
| 6 | |||
| Cu–O2 | 1.940(4) | Cu–O4 | 1.927(4) |
| Cu–O6a | 1.926(4) | Cu–O8a | 1.945(4) |
| Cu–O9 | 2.278(4) | ||
| O2–Cu–O4 | 89.42(16) | O2–Cu–O6a | 89.67(16) |
| O2–Cu–O8a | 177.11(15) | O2–Cu–O9 | 91.64(15) |
| O4–Cu–O6a | 171.63(15) | O4–Cu–O8a | 90.12(17) |
| O4–Cu–O9 | 93.89(15) | O6a–Cu–O8a | 90.37(16) |
| O6a–Cu–O9 | 94.46(15) | O8a–Cu–O9 | 91.23(15) |
| Symmetry operation: a = −x + 1, −y + 1, −z + 1 | |||
| 7 | |||
| Cu–O2 | 1.9426(19) | Cu–O4 | 1.9352(19) |
| Cu–O6a | 1.9320(19) | Cu–O8a | 1.9497(19) |
| Cu–O9 | 2.2658(19) | ||
| O2–Cu–O4 | 88.69(9) | O2–Cu–O6a | 90.31(9) |
| O2–Cu–O8a | 177.12(8) | O2–Cu–O9 | 94.74(8) |
| O4–Cu–O6a | 171.61(8) | O4–Cu–O8a | 90.42(9) |
| O4–Cu–O9 | 96.67(8) | O6a–Cu–O8a | 90.18(9) |
| O6a–Cu–O9 | 91.71(8) | O8a–Cu–O9 | 88.08(8) |
| Symmetry operation: a = −x + 1, −y + 1, −z + 1 | |||
| 8 | |||
| Cu1–O2 | 2.024(2) | Cu1–N1 | 2.018(3) |
| Cu1–N2 | 2.009(3) | Cu1–N3 | 1.982(3) |
| Cu2–N4 | 2.031(3) | Cu2–N4a | 2.031(3) |
| Cu2–N5 | 2.040(3) | Cu2–N5a | 2.040(3) |
| O2–Cu1–N1 | 87.88(12) | O2–Cu1–N2 | 175.24(11) |
| O2–Cu1–N3 | 88.84(11) | N1–Cu1–N2 | 91.71(14) |
| N1–Cu1–N3 | 175.21(13) | N2–Cu1–N3 | 91.85(14) |
| N4–Cu2–N4a | 180.00 | N4–Cu2–N5 | 87.90(14) |
| N4a–Cu2–N5 | 92.10(14) | N4–Cu2–N5a | 92.10(14) |
| N4a–Cu2–N5a | 87.90(14) | N5–Cu2–N5a | 180.00 |
| Symmetry operation: a = −x + 1, −y + 1, −z + 1 | |||
| 9 | |||
| Cu1–O2 | 1.968(5) | Cu1–O6 | 1.960(5) |
| Cu1–N1 | 2.002(6) | Cu1–N2 | 1.986(6) |
| Cu1–O9 | 2.293(6) | Cu2–O4 | 1.966(5) |
| Cu2–O8a | 1.968(5) | Cu2–N3 | 2.019(6) |
| Cu2–N4 | 2.012(6) | Cu2–O10 | 2.290(5) |
| O2–Cu1–O6 | 175.4(2) | O2–Cu1–N1 | 87.4(2) |
| O2–Cu1–N2 | 90.6(2) | O2–Cu1–O9 | 92.95(19) |
| O6–Cu1–N1 | 89.2(2) | O6–Cu1–N2 | 92.1(2) |
| O6–Cu1–O9 | 90.9(2) | N1–Cu1–N2 | 168.2(3) |
| N1–Cu1–O9 | 102.8(2) | N2–Cu1–O9 | 88.9(2) |
| O4–Cu2–O8a | 178.2(2) | O4–Cu2–N3 | 88.1(2) |
| O4–Cu2–N4 | 91.2(2) | O4–Cu2–O10 | 91.44(19) |
| O8a–Cu2–N3 | 92.9(2) | O8a–Cu2–N4 | 87.5(2) |
| O8a–Cu2–O10 | 90.09(19) | N3–Cu2–N4 | 170.7(3) |
| N3–Cu2–O10 | 92.0(2) | N4–Cu2–O10 | 97.2(2) |
| Symmetry operation: a = x + 1, y − 1, z | |||
| 10 | |||
| Cu1–O2 | 1.971(2) | Cu1–O6 | 1.946(2) |
| Cu1–N1 | 2.028(3) | Cu1–N2 | 2.026(3) |
| Cu1–O9 | 2.357(3) | Cu2–O4 | 1.961(2) |
| Cu2–O4a | 1.961(2) | Cu2–N3 | 2.047(3) |
| Cu2–N3a | 2.047(3) | Cu3–O8 | 1.973(2) |
| Cu3–O8b | 1.973(2) | Cu3–N4 | 2.023(3) |
| Cu3–N4b | 2.023(3) | ||
| O2–Cu1–O6 | 169.89(10) | O2–Cu1–N1 | 88.60(10) |
| O2–Cu1–N2 | 95.07(10) | O2–Cu1–O9 | 88.33(9) |
| O6–Cu1–N1 | 85.60(10) | O6–Cu1–N2 | 92.53(10) |
| O6–Cu1–O9 | 84.32(9) | N1–Cu1–N2 | 166.01(12) |
| N1–Cu1–O9 | 98.16(11) | N2–Cu1–O9 | 95.44(11) |
| O4–Cu2–O4a | 180.00 | O4–Cu2–N3 | 92.69(10) |
| O4–Cu2–N3a | 87.31(10) | O4a–Cu2–N3a | 92.69(10) |
| O4a–Cu2–N3 | 87.31(10) | N3–Cu2–N3a | 180.00 |
| O8–Cu3–O8b | 180.00 | O8–Cu3–N4 | 88.44(10) |
| O8b–Cu3–N4b | 88.44(10) | O8–Cu3–N4b | 91.56(10) |
| O8b–Cu3–N4 | 91.56(10) | N4–Cu3–N4b | 180.00 |
| Symmetry operations: a = −x + 1, −y + 2, −z; b = −x + 2, −y + 2, −z + 1 | |||
(a) Complexes containing only Cu, pyromellitate and solvent
Three of our complexes contain only Cu2+, either pm4− or H2pm2−, and H2O (both coordinated and uncoordinated). The first (1) is one of the compounds originally provided by Ken Wade's project. Empirically it is a hexahydrate of Cu2(pm), but the formula is more informatively written Cu2(pm)(OH2)4·2H2O. The same structure has previously been reported,13 but the data for that publication were measured at room temperature, H atoms were not included for any of the water molecules, the final R factor was 0.064, and standard uncertainties are higher, e.g. by a factor of about 3 for Cu–O bond lengths.The copper ion occupies a general position, while the pm4− anion lies on an inversion centre. Copper has a square pyramidal coordination geometry, with two terminal aqua ligands arranged cis in basal positions, carboxylate O atoms of two different pm4− ligands in the other two mutually cis basal positions, and a considerably longer apical bond to a third anion, as shown in Fig. 1; all other Cu⋯O distances are >3 Å. Square pyramidal coordination with a long apical bond is found in most of the structures reported here. Each pm4− ligand is coordinated to six copper centres, with two mutually trans carboxylate groups bonded to one metal each and the other two to two metal ions each, making use of both O atoms. The dihedral angles between carboxylate groups and the benzene ring are two of 30.4 and two of 65.9°, the singly coordinating groups being twisted more out of the ring plane. The copper and pm4− ions generate a polymeric sheet structure, as shown in Fig. 2; this is a space-filling representation with van der Waals radii for the atoms and includes the coordinated aqua ligands but not the uncoordinated water molecules, which lie between the sheets, and it demonstrates that the previous description of this as a 2-dimensional porous structure13 is misleading, there being no significant voids within the layer.
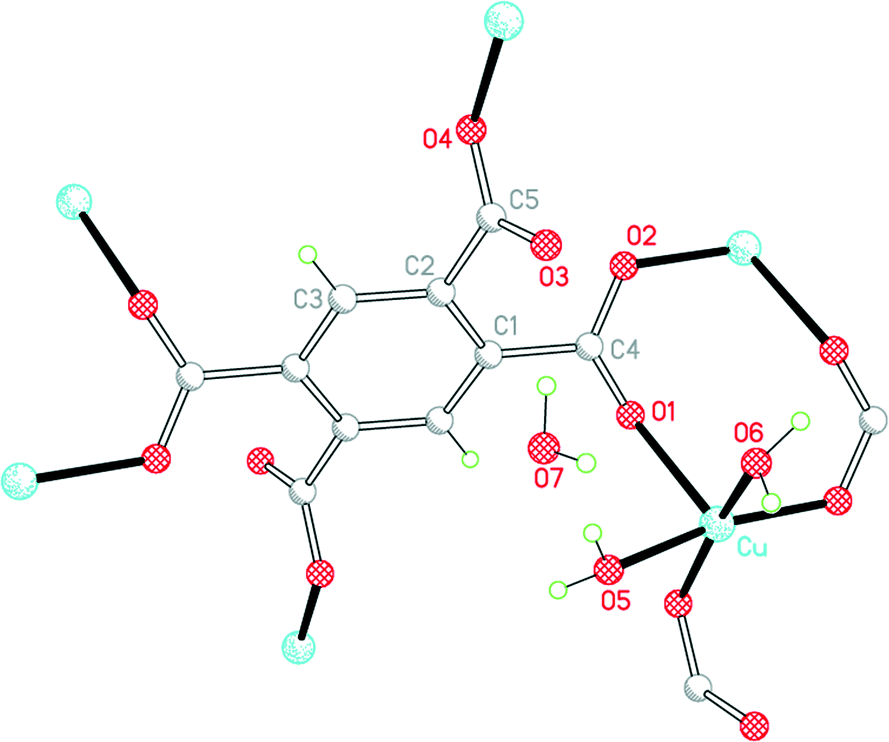 | ||
| Fig. 1 The asymmetric unit of compound 1, with additional atoms to complete the environment of Cu and pm4−. | ||
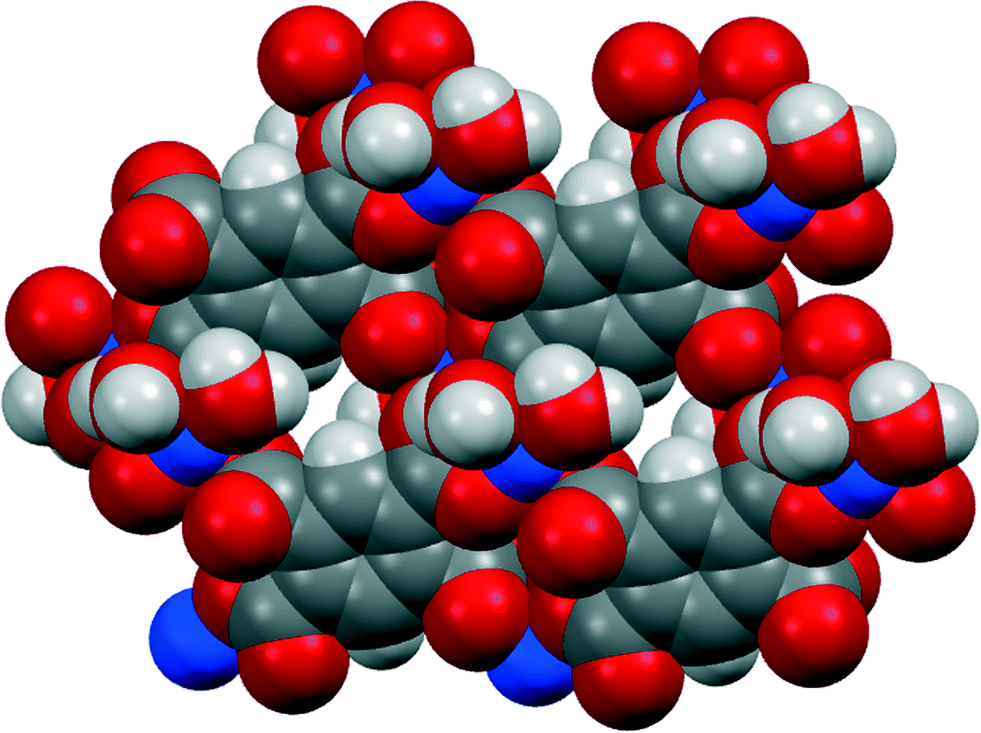 | ||
| Fig. 2 The polymeric sheet structure of compound 1 in a space-filling representation. | ||
Hydrogen bonding involves all six water O–H bonds as donors; the acceptors are all the carboxylate O atoms and the uncoordinated water molecule. A 3-dimensional network is generated, the connections between covalently bonded polymer sheets consisting of direct (aqua)O–H⋯O(pm) interactions as well as interactions via uncoordinated water molecules.
The second structure is that of the higher hydrate Cu2(pm)(OH2)6·4H2O (2), which we have obtained by vapour diffusion of pyridine into a solution of copper(II) nitrate and H4pm in aqueous methanol; for related structures incorporating pyridine as a ligand, see later. It can also be produced by use of triethylamine as base in a gel diffusion experiment. Its synthesis has been reported twice previously, once using potassium hydroxide as base together with H4pm and a copper(II) salt under careful pH control,9,11 and once much more recently by refluxing a mixture of H4pm, CuO and Gd2O3 in water, the role of the gadolinium oxide in the reaction being unclear.14
The 1982 report of this structure was based on room-temperature diffraction data, the R factor is 0.052, and not all H atoms were included; O–H distances range from 0.77 to 1.30 Å. The 2013 report has many disconcerting features: it makes no reference to the earlier structure determination,9 nor to the 2002 publication of compound 1,12 and claims that this is a new compound; it incorrectly states that the pm4− anion lies on an inversion centre instead of a C2 axis; and it claims that the uncoordinated water molecules could not be reliably located and refined, which is surprising given their location in the earlier study, our own straightforward refinement, and the extensive network of hydrogen bonding holding them in place.
The environments of the copper and pm4− ions are shown in Fig. 3. Once again the copper coordination geometry is square pyramidal, but here the apical ligand is aqua and the pairs of aqua and pm4− ligands in the basal plane are each arranged trans. Each carboxylate group of pm4− coordinates one copper ion, though for two of them the second O atom (O3 and its symmetry-equivalent on the adjacent carboxylate) forms a secondary Cu⋯O interaction with length 2.676 Å in the space below the basal plane opposite the apical aqua ligand; all other Cu⋯O distances are >3.2 Å. The dihedral angles between the carboxylate groups and the benzene ring are two of 52.8 and two of 42.1°. Covalently bonded polymeric sheets are formed, which in this case contain substantial spaces surrounded by O and H atoms, as shown in Fig. 4. These are occupied by the uncoordinated water molecules. Contrary to the 2013 description of this structure,14 the water-filled spaces are not continuously connected, in either the a-axis or c-axis direction to form channels, because the successive sheets separated by the c-axis spacing of 9.5816(6)° are interleaved with symmetry-equivalent sheets at half the spacing, which are displaced laterally so that benzene rings of one layer lie above the spaces of the next. Nevertheless it is true that hydrogen bonding, considered alone without the covalent bonding within the polymeric sheets and involving coordinated and uncoordinated water, generates networks that extend along the c axis but do not connect with each other in the other two dimensions. All O–H bonds of the coordinated and uncoordinated water molecules serve as hydrogen-bond donors; the acceptors are all the carboxylate O atoms, the two uncoordinated water molecules, and the apical (but not the basal) aqua ligand.
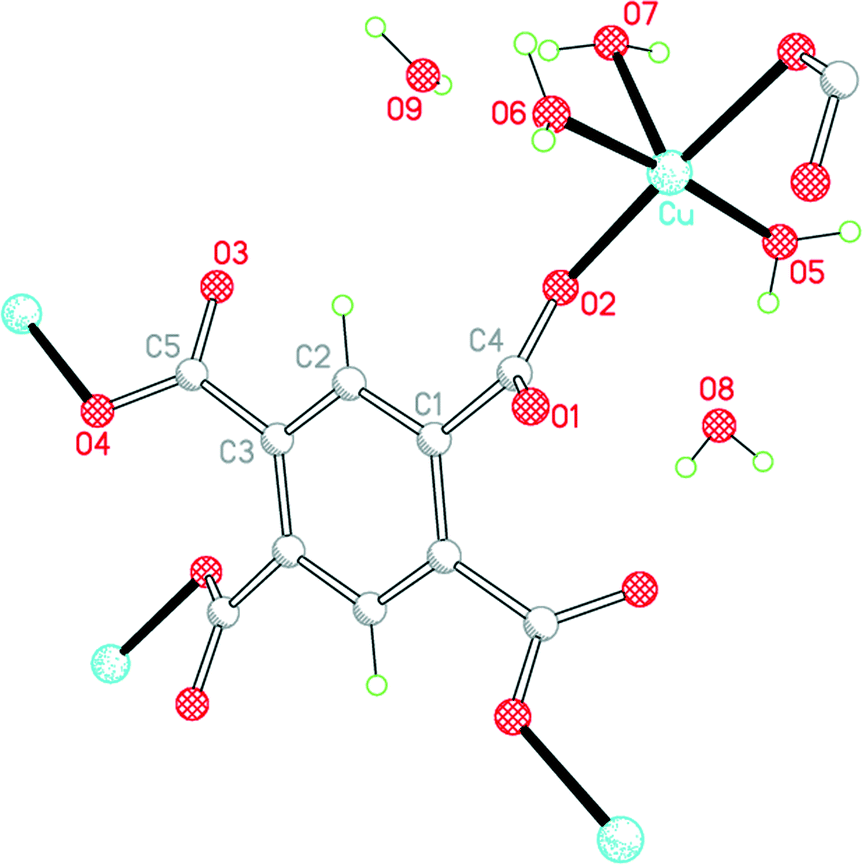 | ||
| Fig. 3 The asymmetric unit of compound 2, with additional atoms to complete the environment of Cu and pm4−. | ||
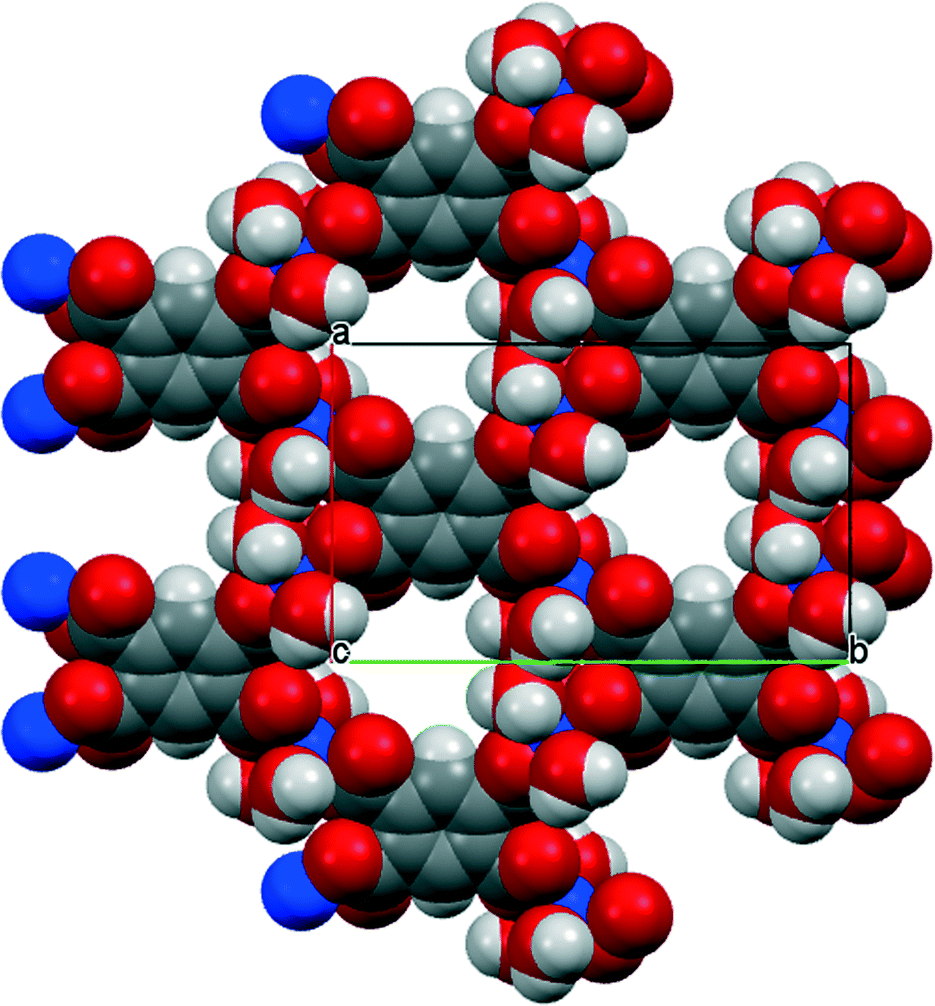 | ||
| Fig. 4 The polymeric sheet structure of compound 2 in a space-filling representation. | ||
Comparison of these two Cu/pm4−/H2O structures shows that the larger proportion of water in 2 means that the five coordination sites of Cu are occupied in the ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 by water and carboxylate ligands instead of 2:3, and there are twice as many uncoordinated water molecules in the structure; concomitantly the connectivity of the pm4− ligand is reduced from 6 to 4 metal centres.
:2 by water and carboxylate ligands instead of 2:3, and there are twice as many uncoordinated water molecules in the structure; concomitantly the connectivity of the pm4− ligand is reduced from 6 to 4 metal centres.
The empirical formula of 3 corresponds to exchange of one Cu2+ in 1 for two protons retained by H2pm2−, maintaining 6 water molecules per anion: Cu(H2pm)(OH2)3·3H2O. All atoms lie in general positions in this, the only structure in the series to have a non-centrosymmetric (but achiral) space group. The two carboxylic acid groups, which are mutually trans, do not coordinate copper, and the two trans-related carboxylate groups act as monodentate donors to copper ions, linking these together into a polymeric chain parallel to the b axis, a segment of which is shown in Fig. 5. The dihedral angles between the substituents and the benzene ring are 84.8 and 86.9° for the coordinating carboxylate groups, and 6.2 and 3.9° for the carboxylic acid groups, which thus achieve near-coplanarity with the ring at the expense of near-orthogonality to the ring for the carboxylate groups.
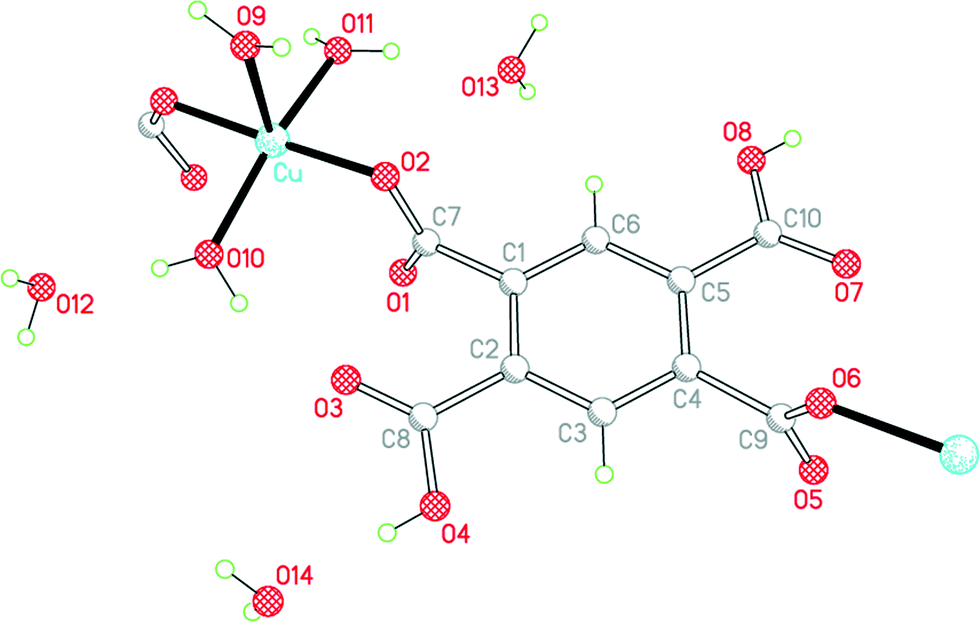 | ||
| Fig. 5 The asymmetric unit of compound 3, with additional atoms to complete the environment of Cu and H2pm2−. | ||
The coordination of copper in 3 is very similar to that in 2 and is best described as square pyramidal, though there is a larger difference between the two basal trans angles in this case. The τ parameter, defined as the difference between these two angles divided by 60°,15 is 0.03 for 2 and 0.20 for 3; it is 0 for ideal square pyramidal, and 1 for ideal trigonal bipyramidal geometry. The structure has been reported twice before,12,16 both times from room-temperature data with R = 0.033 or 0.037 and with H atoms constrained in a riding model without orientational freedom, leading to some poor geometry (both covalent and hydrogen bonding); one of these reports describes the copper coordination as slightly distorted trigonal bipyramidal.12 The next-nearest carboxylate O atom is at a distance of 2.881 Å.
We find, with H atoms refined subject to soft distance restraints, that all 3 coordinated and all 3 uncoordinated water molecules donate their H atoms in hydrogen bonds, as do the two carboxylic acid functions; the acceptors are all 4 carboxylate O atoms, the two carboxylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (but not the carboxylic OH groups), and the 3 uncoordinated water molecules (one of them as a double acceptor), the 3 aqua ligands accepting no hydrogen bonds in this structure. A 3-dimensional hydrogen-bonding network is generated. The shortest hydrogen bonds are between the carboxylic acid OH groups (in which the H atom positions were freely refined without restraints) and one of the water molecules (the double acceptor O14), with O⋯O distances of 2.618(2) and 2.625(3) Å.
O groups (but not the carboxylic OH groups), and the 3 uncoordinated water molecules (one of them as a double acceptor), the 3 aqua ligands accepting no hydrogen bonds in this structure. A 3-dimensional hydrogen-bonding network is generated. The shortest hydrogen bonds are between the carboxylic acid OH groups (in which the H atom positions were freely refined without restraints) and one of the water molecules (the double acceptor O14), with O⋯O distances of 2.618(2) and 2.625(3) Å.
Comparison of the hydrogen bonding in these three structures shows that, when potential donors outnumber acceptors, all donors are used together with all the carboxylate O atoms as acceptors, and uncoordinated water molecules accept hydrogen bonds preferentially over aqua ligands.
There is one other known Cu/pyromellitate/H2O structure containing no other components, the lower hydrate Cu2(pm)(OH2)4, this formula corresponding to that of 1 with all uncoordinated water removed.17 The structure is in marked contrast to those of 1–3, consisting of a 3-dimensional network of copper and pm4− ions in which copper has an almost regular trigonal bipyramidal coordination with terminal aqua ligands in one axial and one equatorial site, the others being occupied by carboxylate O atoms. Hydrogen bonding uses all four O–H bonds of the two aqua ligands, and the acceptors are 3 carboxylate O atoms and one of the aqua ligands. There is also one Cu/pyromellitate/solvent structure in the CSD (two separate reports18) for which the solvent is not water but DMF: Cu2(pm)(DMF)2. This is a 2-dimensional sheet polymer containing ‘paddle-wheel’ units in which a pair of copper ions is bridged by four pm4− ligands in the well-known Cu2(carboxylate)4 arrangement, each Cu has also an apical DMF ligand to give square pyramidal coordination, and each pm4− anion links four such paddle-wheel units to generate the polymer sheet.
(b) Complexes with structures incorporating alkali metal cations
Complexes 1 and 2 have been produced by reactions of copper(II) salts with H4pm and potassium hydroxide in aqueous solution.4,11 The product obtained is dependent on the pH control during the experiment. Complex 4, CuK2(pm)·4H2O, is another product obtainable from this system as the conditions are varied; larger crystals suitable for single-crystal X-ray diffraction were generated by slow diffusion rather than from a single reaction mixture.All components of this structure lie in general positions. This is the only structure in the series for which we have not been able to locate and refine all the water H atoms reliably; one uncoordinated water molecule (O12) has markedly higher displacement parameters than the others and may be subject to incomplete site occupancy, and directions for its O–H bonds to give consistent appropriate hydrogen bonding cannot be found, suggesting that at least the orientation may be disordered. The rest of the structure, however, is completely clear.
In contrast to the first three structures, the primary coordination of copper here is square planar, with carboxylate O atoms as donors. There are two further O atoms 2.601 and 2.611 Å above and below the coordination plane, giving a very distorted octahedral environment with a semi-chelating coordination of two of the carboxylate groups, as shown in Fig. 6.
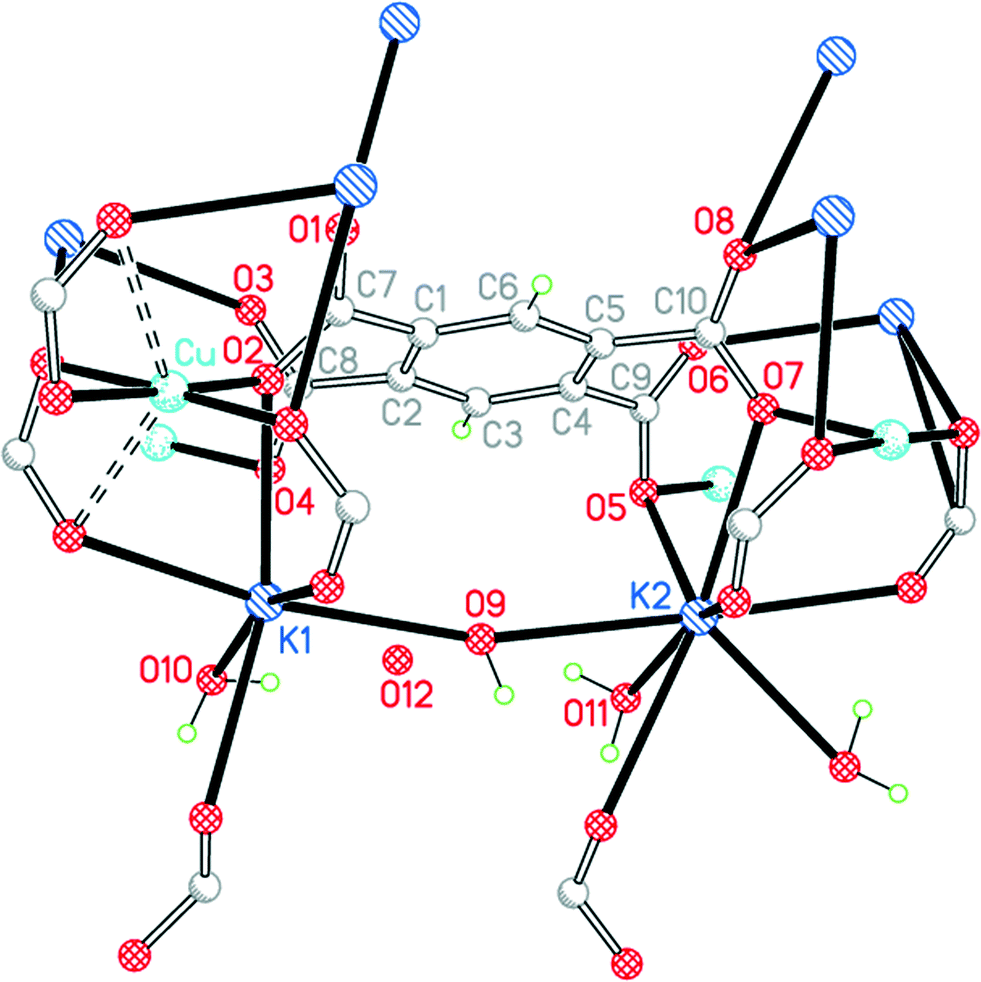 | ||
| Fig. 6 The asymmetric unit of compound 4, with additional atoms to complete the environment of Cu, K1, K2 and pm4−. | ||
The two crystallographically independent potassium cations have different coordination environments. K1 is 7-coordinate (K1–O4 is obscured behind K1–O2 in Fig. 6) with an approximate pentagonal bipyramidal geometry: the axial bonds are to a bridging water molecule O9 and a carboxylate O atom (symmetry-equivalent to O3), and the equatorial plane contains four carboxylate O atoms and the terminal aqua ligand O10. K2 is 8-coordinate with an irregular geometry; it may be regarded as based on 7-coordinate pentagonal bipyramidal with water O9 and a symmetry-equivalent of carboxylate O6 positioned axially, with 3 carboxylate O atoms and a bridging water molecule (O11) in 4 equatorial sites, and with a fifth single equatorial atom replaced by a symmetry-equivalent water O11 and a further carboxylate O atom on either side of the equatorial plane (these are the two bonds to K2 pointing closest to the bottom edge of Fig. 6, and the K–carboxylate bond is significantly longer than the other 7 in this coordination sphere, at 3.216 Å; the next-nearest O atom is at 3.698 Å).
The pm4− ligand coordinates to no fewer than 12 different metal centres, of which 4 are copper, 4 are K1 and symmetry-equivalents, and 4 are K2 and symmetry-equivalents. Each O atom forms two bonds to metal ions, and the carboxylate groups serve both bridging and chelating functions. The dihedral angles between carboxylate groups and the benzene ring are 39.8, 63.3, 58.6 and 42.4°. The copper and pm4− ions alone form a polymeric sheet, and the sheets are connected together through the potassium ions.
The four water molecules all have different functions and environments. O9 is a bridge between K1 and K2 with a wide K–O–K angle of 164.59(12)°. O10 is a terminal ligand for K1. O11 is an asymmetrical bridge between two symmetry-equivalent K2 ions, generating a centrosymmetric parallelogram with two K2 and two O11 as its vertices and K–O–K angles of 102.94(14)°. O12 is not coordinated. O9, O10 and O11 form hydrogen bonds, the acceptors being carboxylate O atoms and O12. Hydrogen bonding in this structure generates discrete clusters of four water molecules surrounded by carboxylate groups.
Complex 5 has the formula CuNa2(pm)·4H2O. Although this is empirically the same as 4 with sodium replacing potassium, the structure is completely different. Cu lies on a C2 axis, Na in a general position, and pm4− on an inversion centre; the sodium and pm4− ions give a covalently connected 3-dimensional framework even without consideration of the sodium ions and water molecules. The pm4− anion is bonded to 4 copper ions (one through each carboxylate group) and to 8 sodium ions. The dihedral angles between the carboxylate groups and the benzene ring are two of 53.9° and two of 56.3°.
As in 4, Cu has a primary square-planar coordination, bonded to four different pm4− ligands, and has one longer Cu⋯O(pm) contact on each side of the plane, both at 2.767 Å, as shown in Fig. 7. Sodium is 6-coordinate in a distorted octahedral geometry, with two cis terminal aqua ligands and four carboxylate O atoms. The two independent water molecules form four hydrogen bonds, the acceptors being three carboxylates and one of the water molecules, generating discrete hydrogen-bonded units of two water molecules and three surrounding carboxylate groups.
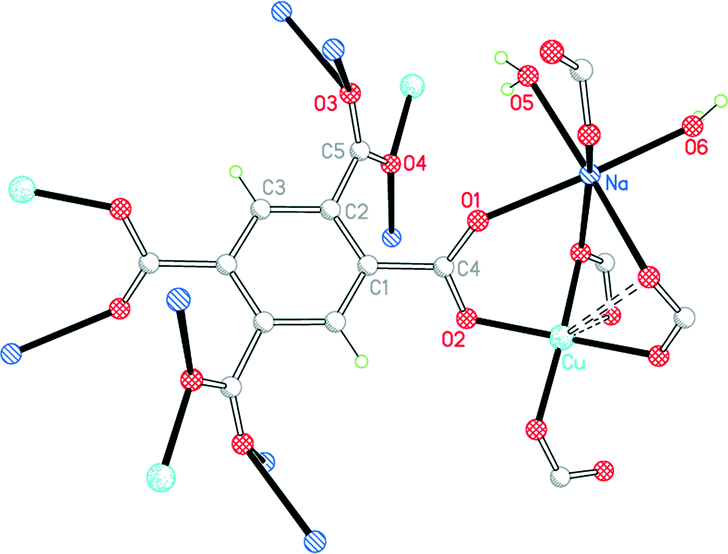 | ||
| Fig. 7 The asymmetric unit of compound 5, with additional atoms to complete the environment of Cu, Na and pm4−. | ||
The important common features of the structures of 4 and 5 are that copper is coordinated only by carboxylate groups, water is coordinated only to the alkali metal ions, and pyromellitate coordinates 12 different cations (4 Cu and 8 alkali metal). By contrast, the other known structure with only Cu, alkali metal, pyromellitate and water as ingredients is CuNa2(H2pm)(OH2)8·2H2O, in which copper is exclusively coordinated by water (4 bridging and 2 terminal in a Jahn–Teller-elongated octahedral geometry with long bonds to two trans bridges), H2pm2− forms only two coordinative bonds and thus bridges two sodium ions, and the structure is a chain polymer with a linear ⋯Na⋯Na⋯Cu⋯ repeat unit;19 the chains are connected by hydrogen bonding, and there is hydrogen bonding also within the completely planar H2pm2− anion.
(c) Anionic complexes with protonated bases as counterions
We have three of these, without literature precedents among copper pyromellitate complexes as far as we are aware, and the structures show some unusual and surprising features.Vapour diffusion of triethylamine into a solution of a copper(II) salt and H4pm in ethanol produces compound 6. The triethylamine deprotonates H4pm and the resulting protonated base serves as counter-cations to the dimeric anion containing Cu, pm4− and ethanol (Fig. 8). Discrete (non-polymeric) metal complexes of pm4− are much less common than polymeric structures, and in most of them coordination sites of the metal centres are blocked by bi- or multidentate ligands that serve as terminators, preventing further association into chains, sheets or networks. The few entries in the CSD for discrete structural units that contain only metal ions, pm4− and coordinated solvent are all neutral dinuclear species in which a single pm4− coordinates through two opposite carboxylate groups to two M(OH2)52+ centres (M = Mg, Mn, Co, Ni).20
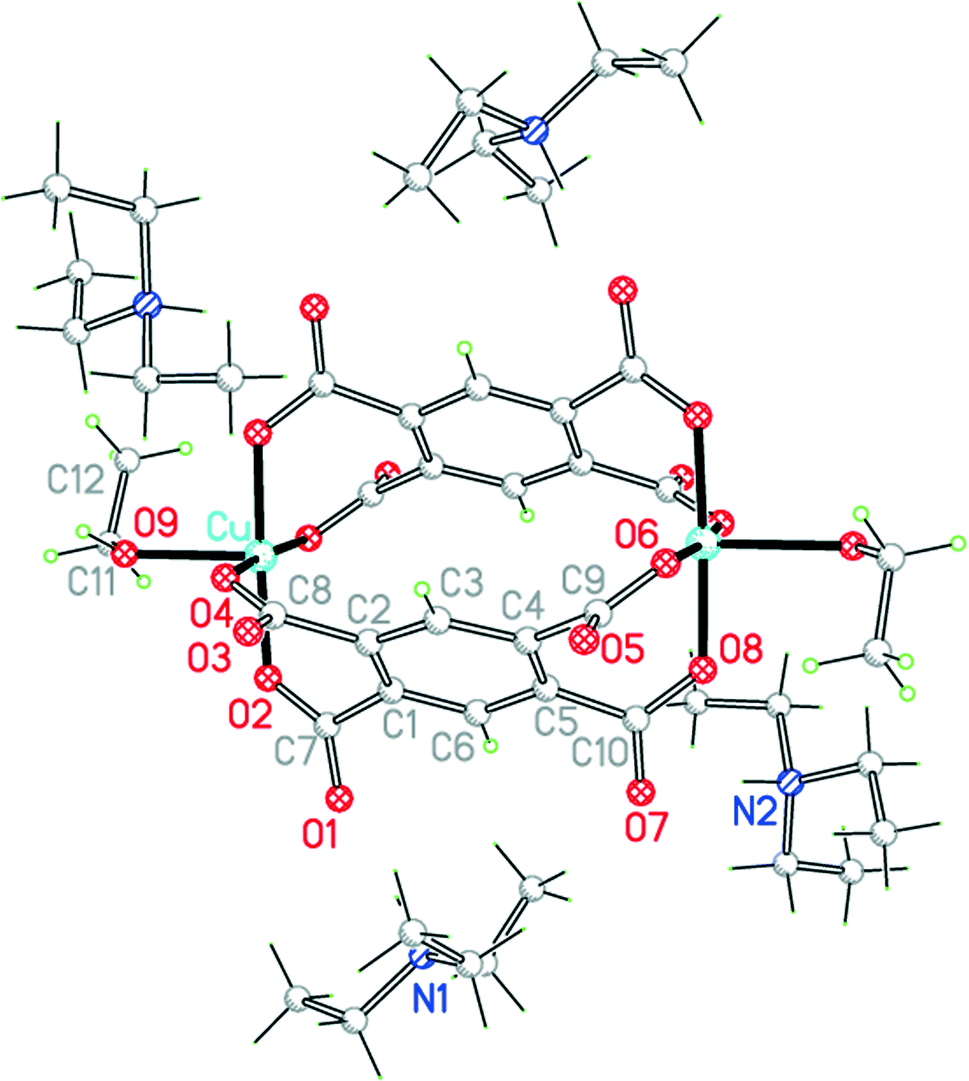 | ||
| Fig. 8 The unit cell contents of compound 6. Minor disorder components are not shown. | ||
The centrosymmetric anion of 6 has two parallel pm4− ligands with their benzene rings separated by 4.155 Å (too far for any significant π-stacking interactions). The carboxylate groups are twisted out of the benzene ring plane by 42.8, 48.2, 56.0 and 39.2°, and the four O atoms lying on one side of the plane coordinate the two copper ions, leaving the other four O atoms protruding on the opposite side and not engaging in coordination. Each pm4− anion acts as a bis-bidentate ligand, adjacent carboxylate groups being attached to the same copper ion. Each of the two copper ions is coordinated in this way by two pm ligands, forming the basal plane in a square pyramidal coordination geometry, the apical position of which is occupied by a molecule of ethanol (with its methyl group disordered over two orientations); the apical bond is considerably longer than the basal bonds, as in the other square pyramidal structures reported here.
Such a ‘lantern’ structure is unprecedentated among metal pyromellitate complexes. There are five previously reported structures in which pm4− coordinates in a bis-bidentate fashion with each pair of adjacent carboxylate groups coordinated to a single metal ion.10,21 In each of these there is only one pm4− ligand and the dinuclear molecule is precisely or approximately centrosymmetric, with the two metal centres lying on opposite sides of the benzene ring.
The ethyl groups of one of the two crystallographically independent Et3NH+ cations are disordered; the N–H groups, however, are ordered and are oriented such as to form N–H⋯O hydrogen bonds to 4 of the 8 uncoordinated carboxylate O atoms of the anion. Further O–H⋯O hydrogen bonds are formed between the ethanol apical ligands and carboxylate O atoms of two adjacent anions, linking the anions together in a hydrogen-bonded chain parallel to the b axis. Two of the uncoordinated O atoms do not serve as hydrogen bond acceptors.
Compound 7, obtained in a similar reaction carried out in methanol rather than ethanol solution, has essentially the same structure with only minor differences (Fig. 9): there is more extensive disorder of one of the cations, methanol shows no disorder, and uncoordinated methanol is incorporated in the crystal structure. The unit cells of the two structures are very similar, with the same space group, and the arrangements of the cations and anions are almost the same, the smaller methanol molecule providing space for extra solvent inclusion that causes a small increase rather than the expected decrease in the unit cell volume. The separation between the two parallel benzene rings is 4.188 Å, and the dihedral angles between the carboxylate groups and the benzene ring within each ligand are 46.4, 49.3, 57.6 and 36.6°. The hydrogen bonding pattern is the same as in compound 6, with the addition that the uncoordinated methanol molecules are hydrogen-bonded to the remaining carboxylate O atoms that do not serve as acceptors in 6.
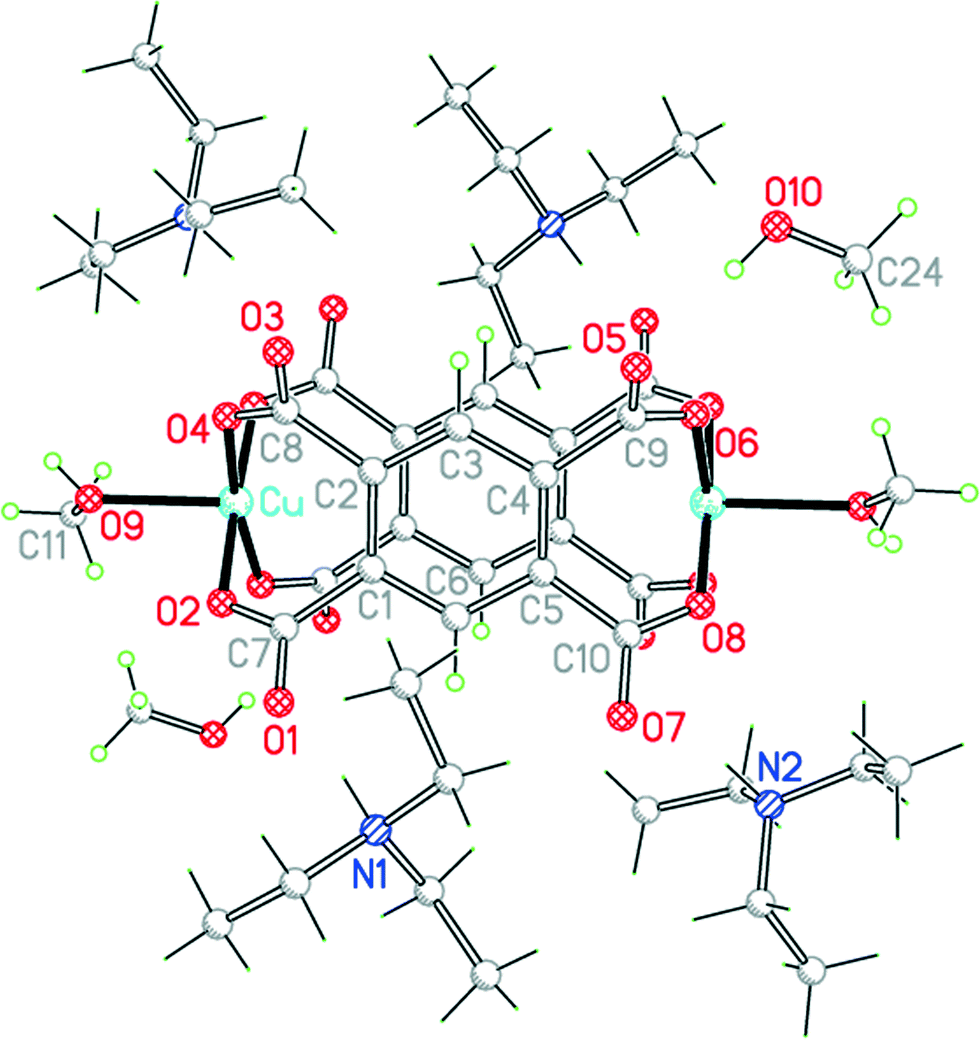 | ||
| Fig. 9 The unit cell contents of compound 7. Minor disorder components are not shown. | ||
Compound 8 results from the use of ammonia as base to deprotonate H4pm; ammonia is incorporated in the structure both as a ligand and as an ammonium cation (Fig. 10). If only primary coordination is considered (bonds to Cu under 2.5 Å in length), the structure contains relatively simple discrete units: NH4+ and centrosymmetric square-planar [Cu(NH3)4]2+ cations, [Cu(pm)(NH3)3]2− anions in which a terminal pm4− ligand replaces one NH3 of the tetraamminecopper(II) cation, and uncoordinated water molecules. As far as we are aware, this is the first occurrence of pm4− as a terminal ligand coordinating through only one O atom. The dihedral angles between the carboxylate groups and benzene ring are 67.2 (coordinated), 18.3, 58.3 and 68.9°.
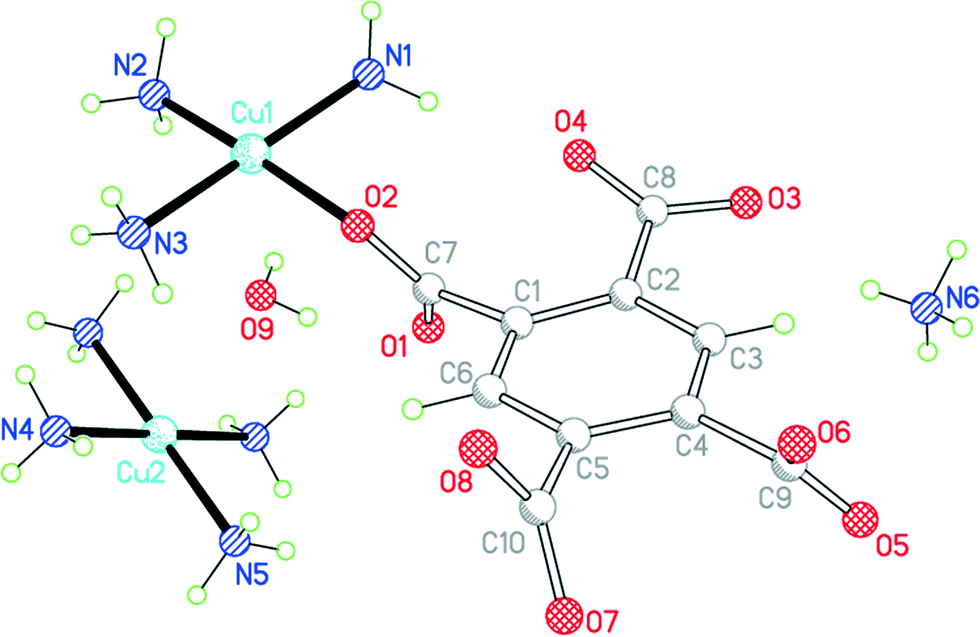 | ||
| Fig. 10 The asymmetric unit of compound 8, with additional atoms to complete the environment of Cu2. | ||
Each of the two copper ions has two O atoms above and below its coordination square plane at distances representing secondary bonding in an elongated octahedral geometry. For Cu2, lying on an inversion centre in the [Cu(NH3)4]2+ cation, the additional ligands are water molecules, with Cu–O = 2.505 Å. A water molecule is also one of the extra ligands of Cu1, at a distance of 2.644 Å; opposite it (O–Cu–O 165.3°) lies a symmetry-equivalent of the coordinating carboxylate atom O2, at 2.581 Å. Two Cu1-containing anions thus form a centrosymmetric pair linked by a Cu2O2 parallelogram with sides of 2.024 and 2.581 Å and angles at O of 106.9°. These Cu⋯OH2 and Cu⋯O(pm) secondary interactions link the various ions (other than ammonium) and water molecules into a polymeric chain in the [101] direction, as shown in Fig. 11.
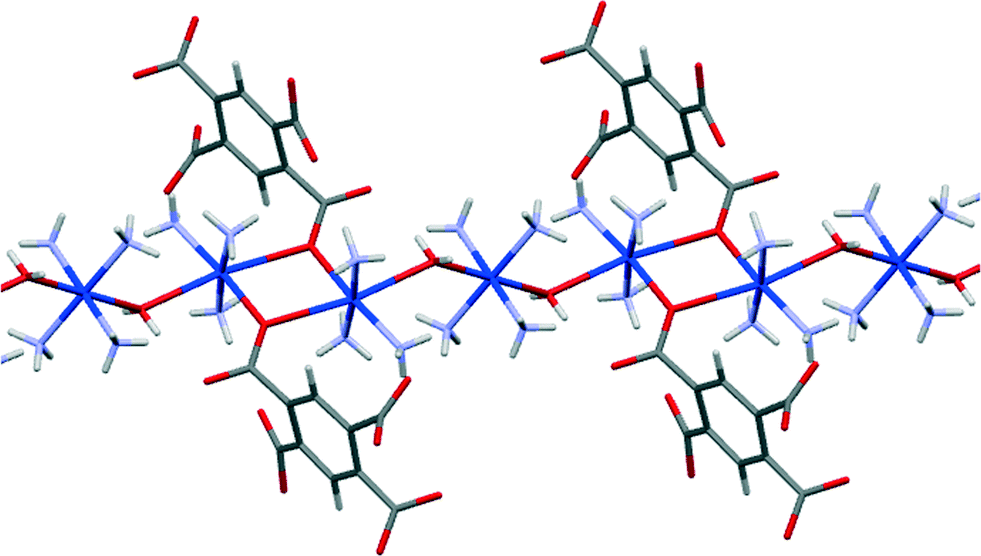 | ||
| Fig. 11 A section of the polymeric chain of compound 8 including secondary coordination bonds. | ||
These chains are connected by an extensive 3-dimensional network of hydrogen bonds, in which all but one of the 19 N–H bonds and both water O–H bonds serve as donors and all O atoms are acceptors.
The only other reported copper pyromellitate complex containing ammonia is the 2-dimensional polymeric Cu2(pm)(NH3)4·2H2O, in which copper has a square pyramidal coordination and each pm4− anion coordinates 6 Cu centres.22 Use of methylamine instead of ammonia generates instead a 3-dimensional polymer Cu2(pm)(NH2Me)4(OH2)2 (note the very similar formula overall) with elongated octahedral Cu coordination, bonding of each pm4− ligand to 4 Cu centres, and water now coordinated as a bridging ligand between pairs of copper ions.
(d) Complexes in which unprotonated bases serve as ligands
The two uncharged polymeric species just mentioned are examples of this class of complex, which is otherwise unrepresented in the literature except for the pyridine-containing Cu2(pm)(py)4(OH2)2·4H2O.23 We have obtained the same compound 9 by a gel diffusion experiment, and a related compound Cu2(pm)(py)4(MeOH)·2MeOH (10) using vapour diffusion of pyridine into a solution in methanol of a copper(II) salt and H4pm. The two structures have many similarities, but there are some significant differences.The structure of 9 has been described, with the main focus on the presence of hydrogen-bonded clusters of 6 water molecules.23 Although the publication states that water H atoms were refined as riding, examination of the deposited CIF suggests that they were actually refined freely. We include our own essentially identical result (of similar precision) here for comparison with the other structures in the series, using consistent refinement treatment and atom labelling schemes (Fig. 12). The asymmetric unit contains two copper ions in general positions, half each of two centrosymmetric pm4− ions (the previous report implies there is a complete pm4− ion without crystallographic symmetry), four coordinated pyridine molecules, two coordinated and four uncoordinated water molecules. The coordination of the two copper ions is the same: square pyramidal with two trans pyridines and two trans carboxylates in the basal plane and a long apical bond to an aqua ligand. Each pm4− ligand coordinates 4 copper ions (two Cu1 and two Cu2), one attached to each carboxylate group in a monodentate fashion. The dihedral angles between the carboxylate groups and the benzene rings are two of 83.2° and two of 6.0° in one ligand, and two of 86.3° and two of 2.8° in the other, a combination of near-coplanarity and near-orthogonality not found in the other complexes in this series except in the H2pm2− ligand of compound 3. Hydrogen bonding is confined to the clusters of 6 water molecules (2 of them being aqua ligands) and 4 surrounding carboxylate groups, described previously, and does not generate a polymeric hydrogen-bonding network.
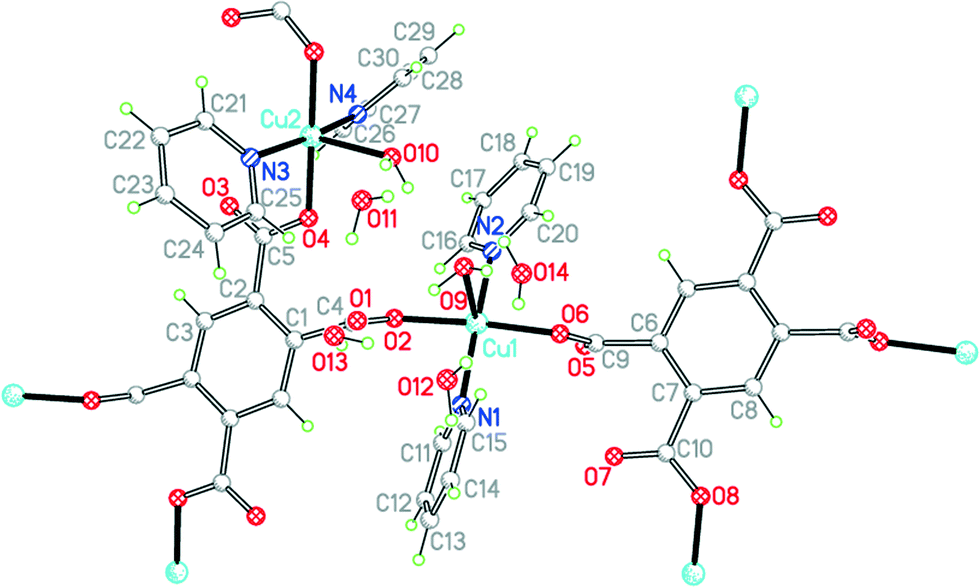 | ||
| Fig. 12 The asymmetric unit of compound 9, with additional atoms to complete the environment of Cu2 and pm4−. | ||
The asymmetric unit of compound 10, in a unit cell of similar volume to that of 9 and the same space group, also contains half each of two centrosymmetric pm4− ligands and four coordinated pyridine molecules, but has three copper ions of which two lie on inversion centres (instead of two in general positions) and three methanol molecules, one of which is clearly a ligand and the other two may be described as semi-coordinating (i.e. half the number of solvent molecules) (Fig. 13). Cu1 has a square pyramidal coordination geometry, similar to that found in compound 9 but with methanol replacing the apical aqua ligand: the basal ligands are two trans pyridines and two trans carboxylates. The next shortest Cu1⋯O distance is 2.781 Å, which may be considered as rendering one carboxylate group semi-chelating. Cu2 and Cu3 both have primary square planar coordination with two trans pyridines and two trans monodentate carboxylates; methanol molecules lie above and below the square plane in each case, at 2.536 and 2.521 Å, thus generating elongated octahedral coordination. As for 9, the Cu and pm4− ions generate 2-dimensional polymer sheets in which pyridine and water or methanol serve only as terminal ligands decorating the sheet. Hydrogen bonding links methanol to carboxylate groups within the sheets and also connects each sheet to those above and below by methanol⋯methanol interactions. Ignoring the Cu1⋯O1 contact, each pm4− ligand has the same connectivity as in 9, with one copper ion attached to each carboxylate group. The dihedral angles between carboxylate groups and benzene rings are two of 55.7° and two of 57.1° for one ligand, and two of 61.7° and two of 31.8° for the other.
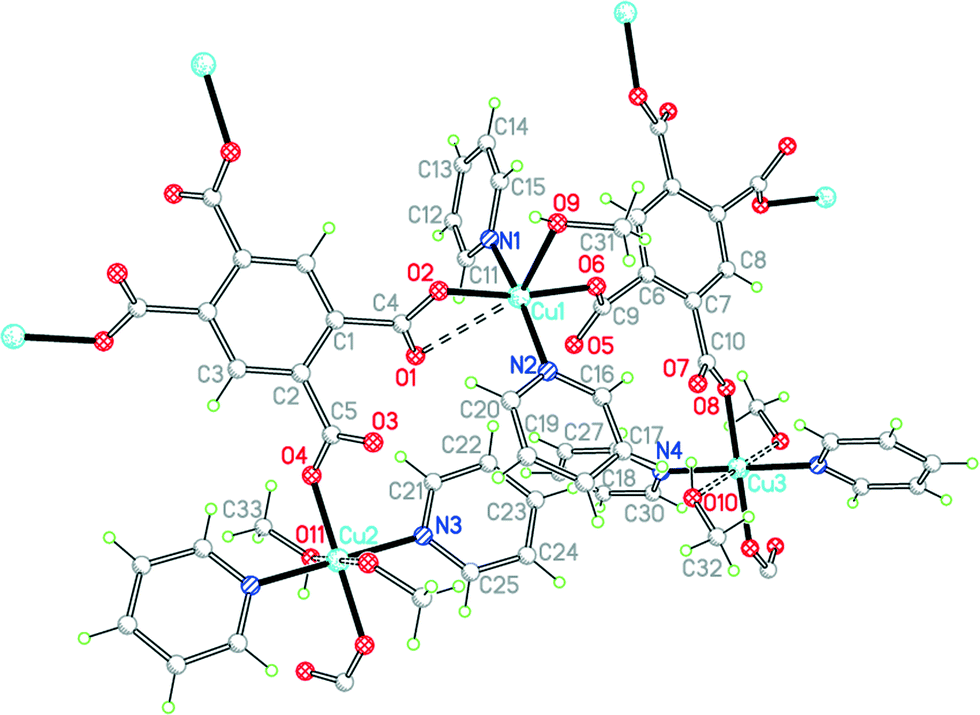 | ||
| Fig. 13 The asymmetric unit of compound 10, with additional atoms to complete the environment of Cu2, Cu3 and pm4−. | ||
Principal comparisons and patterns
In most of the structures described here, the fully deprotonated pyromellitate anion pm4− coordinates 4 or 6 Cu2+ ions, at least one for each carboxylate group, and additionally 8 alkali metal cations in two of the structures. Rarely do carboxylate groups lie anywhere near coplanar with the benzene ring of pyromellitate ligands, most being twisted out of the ring plane by 30–70°. The most common coordination of copper is square pyramidal with a long apical bond, though there are some examples of primary square planar coordination, which is supplemented by two weaker Cu⋯O interactions to give a distorted octahedral geometry. Between 1 and 4 coordination sites are taken by carboxylate groups, the others being occupied by solvent or nitrogen base molecules as ligands. Coordinated solvent molecules are usually terminal ligands, though in some cases water bridges two metal ions. Structures range from discrete molecular ions through chains and sheets to 3-dimensional polymeric networks, sheet polymers being the most common. Hydrogen bonding makes a significant contribution to most of the structures, usually with all potential donors satisfied and a surfeit of acceptors.One particular feature of interest in the 2-dimensional polymeric structures described here concerns the network formed by only the Cu2+ and pm4− ions, comparison of which is facilitated by ignoring all the other components of the structures (other ions, and both coordinated and uncoordinated uncharged molecules). A section of each of the Cu/pyromellitate networks is shown in Fig. 14 (a–e) for compounds 1, 2, 4, 9, and 10, together with Cu2(pm)(OH2)2(MeNH2)4 (f),22 which is similar in this respect. It can be seen that the number of pm4− anions coordinating each copper ion is 2 (linear, b, d, e, f), 3 (pyramidal, a), or 4 (square planar, c), and the number of copper ions coordinated by each pm4− anion is either 4 (b–f) or 6 (a); the ratio of these is 1:2 except for structure 4 (c), where it is 1:1, in accordance with the stoichiometric pyromellitate:Cu ratio of each compound. In each structure, the polymeric network displays a grid of either one unique (b, d, e, f) or equal numbers of two different (a, c) rings made up of alternating copper centres and segments of pm4− ligands. The unique rings contain 4 Cu and 4 pm4− ligands, while the others contain 2 of each, which are smaller. These large rings do not constitute channels or voids in the structures, as they are occupied and surrounded by the counterions, coordinated and uncoordinated molecules that have been omitted from the network diagrams.
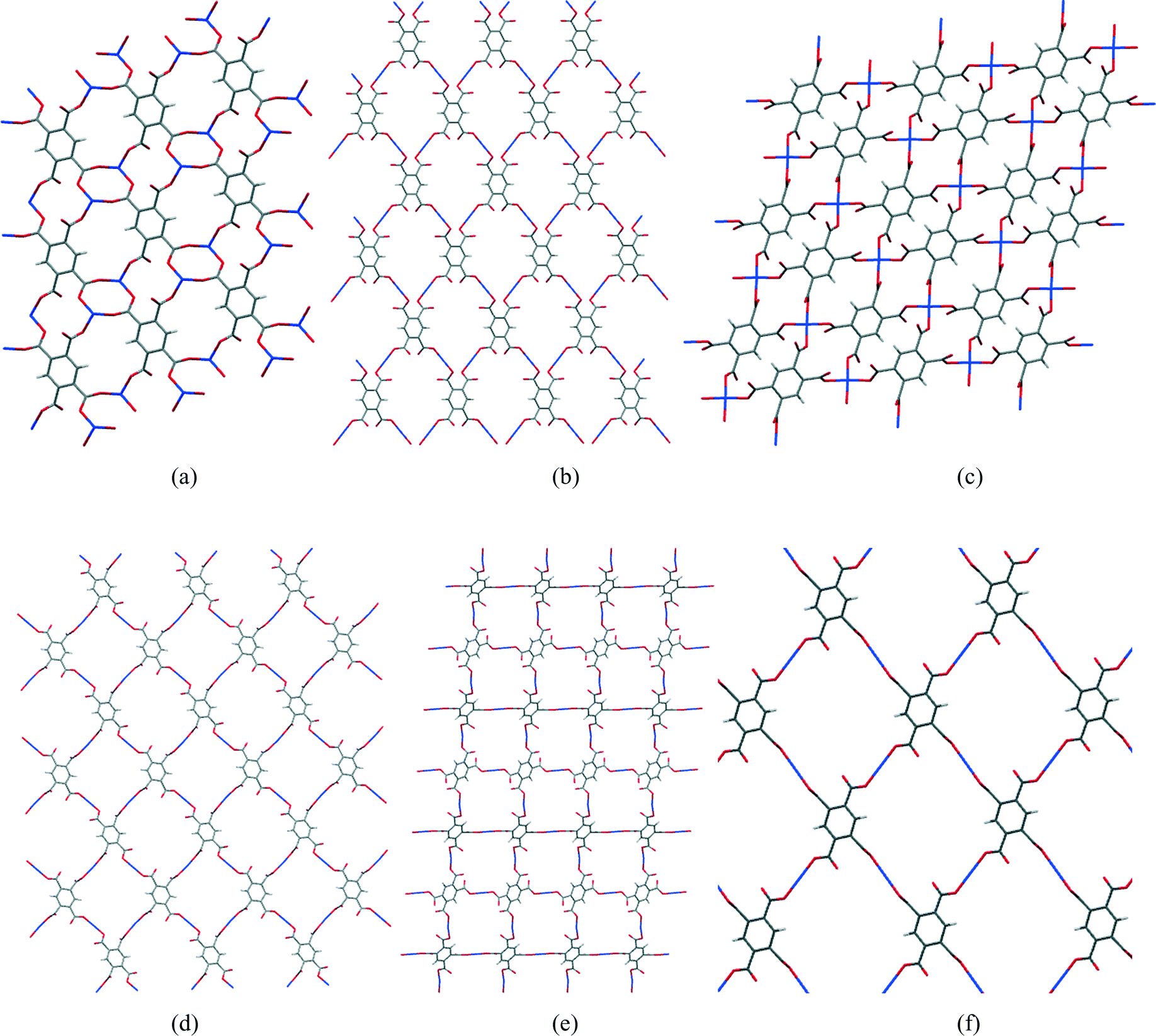 | ||
| Fig. 14 Sections of the 2-dimensional Cu/pm4− nets of compounds 1 (a), 2 (b), 4 (c), 9 (d), 10 (e) and Cu2(pm)(OH2)2(MeNH2)4 (f). | ||
Experimental
Materials and methods
All starting materials were commercial products and were used as supplied; water was distilled, and other solvents were not dried. Chemical analyses were performed with a Carlo Erba 1108 Elemental Analyser in the Newcastle University School of Chemistry Analytical Services Unit. Most syntheses were carried out several times in parallel or on different occasions, using slow diffusion methods in order to obtain single crystals suitable for X-ray diffraction analysis; the methods employed involved either slow diffusion of reactant solutions from opposite sides through a freshly prepared silica gel, diffusion of one solution into a gel containing a non-volatile base, or vapour diffusion of a volatile base into a solution of other reactants, with details given below. In cases where gel synthesis gave more than one product, these were usually generated in different parts of the gel and were collected separately, or crystals of mixed products were separated manually on the basis of optical examination (crystal shape and colour).X-ray crystallography
Crystals were examined and diffraction data collected on three different instruments over a period of several years: a Stoe-Siemens AED2 four-circle diffractometer (1 and 4), an Agilent Technologies Gemini A Ultra diffractometer (10), and a Bruker SMART 1K diffractometer (all others); the radiation was Mo-Kα in each case (λ = 0.71073 Å). For some compounds more than one data set was measured, using crystals from different syntheses and sometimes with different instruments, and the best result is presented here. Selected crystal data are shown in Table 2. Standard diffractometer control software was used, complemented by locally written programs.24 Absorption corrections were based on symmetry-equivalent and repeated reflections measured in different positions and thus subject to different X-ray path lengths.24c,25 The structures were solved and initially refined with SHELXTL;25,26 final refinements used SHELXL-2014.27 Hydrogen atoms were included in idealised positions bonded to C atoms, and refined as riding; most of those bonded to O and N atoms were located in difference density maps and/or assigned to give appropriate hydrogen bonds, and were subsequently refined with restraints on N/O–H and H⋯H distances (providing orientational degrees of freedom) and with displacement parameters tied to those of their parent atoms.| Compound | 1 | 2 | 3 | 4 | 5 |
| Chem formula | C10H2Cu2O8·6H2O | C10H14Cu2O14·4H2O | C10H10CuO11·3H2O | C10H2O8CuK2·4H2O | C10H2CuNa2O8·4H2O |
| Formula mass | 485.29 | 557.35 | 423.77 | 463.92 | 431.70 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic |
| a/Å | 6.512(1) | 12.1518(8) | 6.7846(4) | 7.7391(15) | 6.8940(5) |
| b/Å | 6.901(1) | 18.1413(11) | 10.9535(6) | 10.6004(19) | 16.3377(11) |
| c/Å | 9.328(2) | 9.5816(6) | 10.6835(6) | 10.6159(19) | 12.9381(9) |
| α/° | 95.448(10) | 90 | 90 | 112.867(10) | 90 |
| β/° | 108.706(8) | 113.503(2) | 91.810(2) | 90.411(13) | 99.666(2) |
| γ/° | 108.090(8) | 90 | 90 | 104.192(10) | 90 |
| V/Å3 | 368.51(11) | 1937.0(2) | 793.55(8) | 772.8(3) | 1436.56(17) |
| T/K | 240(2) | 160(2) | 160(2) | 295(2) | 160(2) |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c | Pn |
P |
C2/c |
| Z | 1 | 4 | 2 | 2 | 4 |
| Reflections measured | 2592 | 7999 | 6540 | 5452 | 6058 |
| Independent reflections | 1296 | 2253 | 3467 | 2726 | 1732 |
| R int | 0.0262 | 0.0258 | 0.0173 | 0.0265 | 0.0242 |
| R 1 (I > 2σ) | 0.0260 | 0.0235 | 0.0191 | 0.0285 | 0.0205 |
| wR(F2) (I > 2σ) | 0.0688 | 0.0593 | 0.0484 | 0.0789 | 0.0564 |
| R 1 (all data) | 0.0270 | 0.0266 | 0.0199 | 0.0360 | 0.0208 |
| wR(F2) (all data) | 0.0700 | 0.0608 | 0.0488 | 0.0837 | 0.0565 |
| GoF on F2 | 1.092 | 1.084 | 1.058 | 1.062 | 1.157 |
| Compound | 6 | 7 | 8 | 9 | 10 |
| Chem formula | 4C6H16N+·C24H16Cu2O184− | 4C6H16N+·C22H12Cu2O184−·2CH4O | 2NH4+·CuN4H122+·2C10H11CuN3O82−·2H2O | C30H26Cu2N4O10·4H2O | C33H34Cu2N4O11 |
| Formula mass | 1128.24 | 1164.27 | 933.31 | 801.69 | 789.72 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Triclinic | Triclinic |
| a/Å | 11.3114(10) | 11.5053(11) | 8.2343(7) | 8.9464(9) | 8.7980(3) |
| b/Å | 11.5464(10) | 11.5543(11) | 19.5788(16) | 11.1622(11) | 10.6480(4) |
| c/Å | 11.7074(11) | 12.0160(11) | 10.9336(9) | 17.4780(17) | 19.1470(8) |
| α/° | 80.130(2) | 78.671(2) | 90 | 82.360(2) | 94.840(3) |
| β/° | 66.441(2) | 64.251(2) | 100.371(2) | 81.293(2) | 90.453(3) |
| γ/° | 71.052(2) | 71.747(2) | 90 | 82.365(2) | 110.355(4) |
| V/Å3 | 1324.1(2) | 1362.9(2) | 1733.9(2) | 1698.6(3) | 1674.37(12) |
| T/K | 160(2) | 160(2) | 160(2) | 160(2) | 150(2) |
| Space group |
P |
P |
P21/c |
P |
P |
| Z | 1 | 1 | 2 | 2 | 2 |
| Reflections measured | 7999 | 11335 | 11979 | 12220 | 13658 |
| Independent reflections | 4562 | 6487 | 3952 | 5926 | 6640 |
| R int | 0.0493 | 0.0142 | 0.0429 | 0.0873 | 0.0391 |
| R 1 (I > 2σ) | 0.0735 | 0.0520 | 0.0451 | 0.0638 | 0.0451 |
| wR(F2) (I > 2σ) | 0.1753 | 0.1342 | 0.0904 | 0.1210 | 0.0963 |
| R 1 (all data) | 0.1086 | 0.0588 | 0.0651 | 0.1394 | 0.0750 |
| wR(F2) (all data) | 0.1927 | 0.1368 | 0.0981 | 0.1489 | 0.1039 |
| GoF on F2 | 0.984 | 1.055 | 1.094 | 1.081 | 1.016 |
Synthetic procedures
Compound 1.4 The copper hydroxynitrate Gerhardtite, Cu2(NO3)(OH)3 prepared from potassium carbonate and copper(II) nitrate (0.649 g, 2.6 mmol), was added to a hot aqueous solution of H4pm (0.792 g, 2.7 mmol in 45 ml); carbon dioxide was evolved and the solution changed from light green to turquoise. The resulting suspension was refluxed for 1 h, to give a blue crystalline solid. Calc. for C10H14Cu2O14: C 24.8, H 2.9; found: C 25.1, H 2.8%.Compound 2. Solutions were prepared of Cu(NO3)2·4H2O (0.079 g, 0.33 mmol) in methanol (10 ml) and H4pm (0.043 g, 0.17 mmol) in water (10 ml). A small sample vial containing 0.5 ml of each solution was almost sealed with plastic film and placed inside a larger closed vial containing pyridine (0.02 ml, 0.11 mmol). Blue block crystals formed after a few days. Calc. for C10H22Cu2O18: C 21.6, H 4.0; found: C 21.7, H 4.3%.
Compound 3. Silica gel was prepared from tetramethoxysilane (5%) and water and allowed to set for 24 hours. On one side of the gel was placed a solution of H4pm (0.042 g, 0.17 mmol) and CuCl2·6H2O (0.045 g, 0.33 mmol) in methanol (1 ml), and on the other triethylamine (0.67 ml). Blue crystals formed over a period of two weeks. Calc. for C10H16CuO14: C 28.3, H 3.8; found: C 28.6, H 3.6%.
Compound 4.11 Aqueous solutions of CuSO4·5H2O and K4pm (prepared from KOH and H4pm in 4:1 molar ratio) were allowed to diffuse together slowly through a fine pyrex sinter, giving blue crystals. Calc. for C10H10CuK2O12: C 25.9, H 2.2; found: C 26.0, H 2.5%.
Compound 5. A 10% silica gel was prepared, containing sodium carbonate (0.212 g, 2 mmol) and H4pm (0.254 g, 1 mmol) in 5 ml water, and allowed to set for 24 hours. CuCl2·6H2O (0.134 g, 1 mmol) in 1 ml ethanol was added on top of the gel. Blue crystals formed in three weeks. Calc. for C10H10CuNa2O12: C 27.8, H 2.3; found: C 27.5, H 2.1%.
Compound 6. A solution of CuCl2·6H2O (0.045 g, 0.33 mmol) and H4pm (0.042 g, 0.17 mmol) in ethanol (3 ml) was placed in a small sample vial, which was almost sealed with plastic film and placed inside a larger closed vial containing triethylamine (0.67 ml). Blue crystals formed in several days. Calc. for C48H80Cu2N4O18: C 51.1, H 7.2, N 5.0; found: C 50.7, H 7.7, N 4.9%.
Compound 7. The same method and quantities were used as for compound 6, but with methanol instead of ethanol as the solvent. A few blue crystals formed in several days, embedded in a pale blue pasty precipitate of heterogeneous appearance; there was insufficient product for reliable chemical analysis.
Compound 8. A solution of CuCl2·6H2O (0.045 g, 0.33 mmol) and H4pm (0.042 g, 0.17 mmol) in methanol (2.5 ml) was placed in a small sample vial, which was almost sealed with plastic film and placed inside a larger closed vial containing 1M aqueous ammonia (1 ml). Blue-purple crystals formed in several days. Calc. for C20H46Cu3N12O18: C 25.7, H 5.0, N 18.0; found: C 26.0, H 4.7, N 17.7%.
Compound 9. Silica gel was prepared from tetramethoxysilane (5%) and water and allowed to set for 24 hours. On one side of the gel was placed a solution of H4pm (0.042 g, 0.17 mmol) and CuCl2·6H2O (0.045 g, 0.33 mmol) in ethanol (2.5 ml), and on the other pyridine (1 ml). Blue crystals formed over a period of two weeks. Calc. for C30H34Cu2N4O14: C 44.9, H 4.3, N 7.0; found: C 44.9, H 4.0, N 6.8%.
Compound 10. Solutions were prepared of Cu(NO3)2·4H2O (0.079 g, 0.33 mmol) in methanol (10 ml) and H4pm (0.043 g, 0.17 mmol) in methanol (10 ml). A small sample vial containing 0.5 ml of each solution was almost sealed with plastic film and placed inside a larger closed vial containing pyridine (0.05 ml, 0.22 mmol). Blue block crystals formed after a few days. Calc. for C33H34Cu2N4O11: C 50.2, H 4.3, N 7.1; found: C 49.9, H 4.1, N 6.8%.
Conclusions
The pyromellitate anion displays considerable versatility in its formation of complexes with copper(II), as a result of the availability of four carboxylate groups, each capable of coordinating one or more metal ions in a variety of modes, and the flexibility of orientation of the carboxylate groups relative to the benzene ring and to each other, subject to steric restrictions. While copper(II) shows a preference for square pyramidal coordination in these complexes, it can also adopt a square planar geometry, usually with additional secondary coordination on each side of this plane. Structural details are susceptible to a balance of influences of other uncharged ligands, including solvent, of organic or alkali metal cations providing overall charge balance, and of hydrogen bonding. Since markedly different structures can be generated from the same or similar ingredients under broadly similar conditions, and the same structure is sometimes found for products from quite different synthetic routes, rational design is unreliable as a guide in benzenepolycarboxylate metal complexes; we have come to the same conclusion based on a series of results for metal mellitate complexes.7 Nevertheless, the research area provides much interest and the opportunity for recognition of patterns and some rationalization of the fascinating structures it generates.Acknowledgements
We thank EPSRC and its predecessor SERC for funding (studentship for NCM and three equipment grants). Compounds 6 and 7 were first prepared and the structures solved as part of an undergraduate research project of Colette Harvey at Newcastle, jointly supervised by Dr Mark Elsegood (now at Loughborough University). Compounds 1 and 4 were provided by Drs Hugh MacBride and Stan Hauptman working with Professor Ken Wade at Durham University, and their experimental procedures are included here. We also thank Drs Mark Fox (Durham) and Gavin Walker (now at Nottingham University) for additional information, including the PhD thesis of Gavin Walker.Notes and references
- (a) W. Clegg, R. Snaith, H. M. M. Shearer, K. Wade and G. Whitehead, J. Chem. Soc., Dalton Trans., 1983, 1309 RSC; (b) D. Barr, W. Clegg, R. E. Mulvey, R. Snaith and K. Wade, J. Chem. Soc., Chem. Commun., 1986, 295 RSC; (c) D. R. Armstrong, D. Barr, W. Clegg, R. E. Mulvey, D. Reed, R. Snaith and K. Wade, J. Chem. Soc., Chem. Commun., 1986, 869 RSC; (d) D. R. Armstrong, D. Barr, R. Snaith, W. Clegg, R. E. Mulvey, K. Wade and D. Reed, J. Chem. Soc., Dalton Trans., 1987, 1071 RSC.
- W. Clegg, Crystal Structure Determination, Oxford University Press, 1998 (2nd edition to be published under the title X-Ray Crystallography, May 2015) Search PubMed.
- W. Clegg, W. R. Gill, J. A. H. MacBride and K. Wade, Angew. Chem., Int. Ed. Engl., 1993, 32, 1328 CrossRef.
- G. S. Walker, PhD thesis, University of Durham, 1995, http://etheses.dur.ac.uk/5312/ Search PubMed.
- (a) J. Kim, B. Chen, T. M. Reineke, H. Li, M. Eddaoudi, D. B. Moler, M. O'Keefe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 8239 CrossRef CAS PubMed; (b) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 649 CrossRef PubMed; (c) A. L. Grzesiak, F. J. Uribe, N. W. Ockwig, O. M. Yaghi and A. J. Matzger, Angew. Chem., Int. Ed., 2006, 45, 2553 CrossRef CAS PubMed.
- W. Clegg and L. Russo, Cryst. Growth Des., 2009, 9, 1158 CAS.
- W. Clegg and J. M. Holcroft, Cryst. Growth Des., 2014, 14, 6282 CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2002, 58, 380 CrossRef.
- B. T. Usubaliev, A. N. Shnulin and K. S. Mamedov, Koord. Khim., 1982, 8, 1532 CAS.
- P. Chaudhuri, K. Oder, K. Wieghardt, S. Gehring, W. Haase, B. Nuber and J. Weiss, J. Am. Chem. Soc., 1988, 110, 3657 CrossRef CAS.
- J. A. H. MacBride, Private communication, 1989 Search PubMed.
- R. Diniz, H. A. de Abreu, W. B. de Almeida, M. T. C. Sansiviero and N. G. Fernandes, Eur. J. Inorg. Chem., 2002, 1115 CrossRef CAS.
- R. Cao, Q. Shi, D. Sun, M. Hong, W. Bi and Y. Zhao, Inorg. Chem., 2002, 41, 6161 CrossRef CAS PubMed.
- P. Cancino, E. Spodine, V. Paredes-García, D. Venegas-Yazigi and A. Vega, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1344 CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- J.-Y. Guo, T.-L. Zhang, J.-G. Zhang, X.-J. Qiao, L. Yang, W. Yu and R.-F. Wu, Wuji Huaxue Xuebao, 2006, 22, 2179 CAS.
- Z. Ling, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m3190 CAS.
- (a) H.-K. Zhao, B. Ding, E.-C. Yang, X.-G. Wang and X.-J. Zhao, Z. Anorg. Allg. Chem., 2007, 633, 1735 CrossRef CAS; (b) A. F. Jalbout, X.-H. Li, M. R. Hassan and G. M. G. Hossain, Transition Met. Chem., 2008, 33, 597 CrossRef CAS.
- Z.-X. Du, J.-X. Li and J.-H. Qin, Z. Kristallogr. - New Cryst. Struct., 2008, 223, 105 CAS.
- (a) D. Cheng, Y. Zheng, J. Lin, D. Xu and Y. Xu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 523 Search PubMed; (b) F. D. Rochon and G. Massarweh, Inorg. Chim. Acta, 2000, 304, 190 CrossRef CAS; (c) F. D. Rochon and G. Massarweh, Inorg. Chim. Acta, 2001, 314, 163 CrossRef CAS; (d) M.-T. Ding, J.-Y. Wu, Y.-H. Liu and J.-L. Lu, Inorg. Chem., 2009, 48, 7457 CrossRef CAS PubMed; (e) H.-K. Liu, T.-H. Tsao, C.-H. Lin and V. Zima, CrystEngComm, 2010, 12, 1044 RSC.
- (a) Z. Shi, J. Jin, L. Zhao, P. Chen and S. Niu, Chin. Sci. Bull., 2010, 55, 124 CrossRef CAS; (b) C.-K. Xia, C.-Z. Lu, D.-Q. Yuan, Q.-Z. Zhang, X.-Y. Wu, S.-C. Xiang, J.-J. Zhang and D.-M. Wu, CrystEngComm, 2006, 8, 281 RSC; (c) C. Zhuang, N. Li and X.-Y. Yu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m268 CAS; (d) Z. Shi, J. Jin, S. Niu, L. Zhang, L. Li and Y. Chi, Huaxue Xuebao, 2009, 67, 2087 CAS.
- Y. Y. Karabach, A. M. Kirillov, M. Haukka, J. Sanchiz, M. N. Kopylovich and A. J. L. Pombeiro, Cryst. Growth Des., 2008, 8, 4100 CAS.
- S. K. Ghosh and P. K. Bharadwaj, Inorg. Chem., 2004, 43, 5180 CrossRef CAS PubMed.
- (a) STADI4, Stoe & Cie GmbH, Darmstadt, Germany, 1985–1990 Search PubMed; (b) SMART and SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 1994–2008 Search PubMed; (c) CrysAlisPro, Agilent Technologies Ltd. (formerly Oxford Diffraction), Oxford, UK, 2008–2011 Search PubMed.
- SHELXTL and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 1994–2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
Footnotes |
| † Dedicated to the memory of Ken Wade, who first introduced me to the beauty of benzenepolycarboxylate chemistry, and who valued precise structural results and used them with such enormous impact. |
| ‡ Electronic supplementary information (ESI) available: Full formatted tables of crystallographic results. CCDC 1044116–1044125 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce00470e |
| § Current address: School of Chemical Engineering and Advanced Materials, Newcastle University, Newcastle upon Tyne NE1 7RU, UK. |
| ¶ Current address: Natrium Capital Limited, The Euston Office, One Euston Square, 40 Melton Street, London NW1 2FD, UK. |
| This journal is © The Royal Society of Chemistry 2015 |