DOI:
10.1039/C5CE00180C
(Paper)
CrystEngComm, 2015,
17, 2737-2753
Crystalline inclusion properties of new pyridine and thiophene modified wheel-and-axle diol hosts†
Received
26th January 2015
, Accepted 24th February 2015
First published on 26th February 2015
Abstract
Structural extension of the ‘wheel-and-axle’ host concept has given rise to the design of a new type of host derivative (1–3) featuring two di(pyrid-2-yl)hydroxymethyl or di(thien-2-yl)hydroxymethyl moieties attached to a central phenylene unit, i.e. having the phenyl groups of a well-known model compound (Ph) substituted by heteroaromatics. Syntheses of the new derivative compounds are described, and the capability to form crystalline inclusions obtained from solvent solution compared to the prototype molecule is studied, including X-ray diffraction analysis of relevant crystal structures. Sorption measurements of the compounds as solid receptor films coated on a quartz crystal microbalance considering a variety of solvent vapors have been performed, showing potential application as mass sensitive sensor materials.
Introduction
Due to their potential uses in a variety of fundamental and practical issues such as compound separation and storage,1–4 crystalline inclusion compounds5–8 are a very challenging field in supramolecular chemistry9 including crystal engineering.10–12 This encouraged the development of appropriate host molecules that are effective in the corresponding behavior patterns.13,14 With reference to the different lines of structural design, hosts based on coordinato-clathrate15,16 and wheel-and-axle strategies17,18 are perhaps the most useful. A prototype host molecule derived from this line of thought, featuring two bulky diphenylhydroxymethyl moieties attached at positions 1 and 4 to a phenylene unit, is given with the formula Ph in Fig. 1.19,20 Not only this particular compound but also its derivatives, showing a variety of substituents, are distinguished by a wide range of inclusion formation properties.21 Nevertheless, notwithstanding some few related structures of heteroaromatic group containing diols possessing, however, the heterocycle in the axle position or that differ in the axle element,22 concrete analogues of Ph showing the phenyl residues substituted by heterocycles such as pyridyl or thienyl units have not been described so far.23–25 Actually, this is a remarkable fact since, as a consequence of the replacement, a change in the crystal packing and specifically modified inclusion behavior might be expected. As for that, this should especially apply to the pyridino derivative of Ph providing additional acceptor sites for hydrogen bonding by means of the pyridine nitrogens. On the other hand, pyridyl and thienyl are spatially very similar to phenyl (isosterism),26 and beyond that, in the latter case, also concerning electronics being, e.g., expressed by the finding that an exchange of phenyl for thienyl in a drug molecule mostly conserves the pharmaceutical effect (bioisosterism).27 In order to prove the above presumptions, the corresponding pyridyl and thienyl derivatives of Ph (Fig. 1), i.e.1 and 2, respectively, were synthesized and comparatively studied with reference to the formation of crystalline inclusion compounds. Considering a structural modification of 1, compound 3 (Fig. 1) featuring a 1,3- instead of a 1,4-connected phenylene unit has been included in the respective study. Furthermore, we give a detailed report on the relevant crystal structures of the corresponding solvent inclusions that have been isolated (1a, 1b, 2a and 2b) and of the solvent-free compounds (1 and 3) with the latter existing in three different polymorphs (3A–3C). Finally, the results of vapor sorption using a quartz crystal microbalance (QCM)28 with 1 and 2 are discussed, and conclusions are drawn from all the findings.
 |
| | Fig. 1 Compounds studied in this paper (for the purpose of comparison, Ph is designated as the known wheel-and-axle host analogue). | |
Results and discussion
Preparation of compounds
Diols 1 and 3 were synthesized from 2-pyridyl lithium (obtained from 2-bromopyridine and n-butyl lithium in dry THF at −78 °C, under argon) and terephthaloyl or isophthaloyl chloride, respectively, yielding the compounds as beige solids. Imitating a described procedure,29 diol 2 was prepared by reaction of 2-thienyl lithium (from thiophene and n-butyl lithium in dry THF at −30 °C, under argon) and dimethyl terephthalate, resulting in a white solid. Inclusion compounds 1a, 1b, 2a, and 2b were obtained by recrystallization of 1 and 2, respectively, from the respective solvents.
Capability of inclusion formation
To investigate the potency of solids 1–3 as hosts for crystalline inclusion formation, they were crystallized from a variety of solvents ranging from dipolar protic via dipolar aprotic to largely apolar species and involving solvents of acyclic and cyclic as well as of aliphatic and aromatic or heteroaromatic nature. Both specification of the solvents and findings obtained from this study are summarized in Table 1; previous results for Ph are also included for comparison.19–21 The most remarkable observation is that under the given conditions, the 1,3-connected pyridino diol 3 totally failed in the inclusion formation and is thus excluded in the table. However, crystallization of 3 from different solvents or work-up of reaction product yielded three polymorphous crystals (3A–3C) of which one (3A) has been isolated only as an irreproducible result, which is discussed in more detail below. On the other hand, the formation of solvent inclusions of the 1,4-connected pyridino and thiopheno diols 1 and 2, respectively, was successful although, compared to the efficiency of Ph, highly limited in number. Particularly, this is the case of 1 giving only stoichiometric 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 inclusions with 1,4-dioxane (1a) and toluene (1b). Hence, it seems to be clear that 1 is only effective in inclusion formation with apolar solvents, and also one should think that the pyridine nitrogens are not involved in a specific host–guest interaction but are possibly used up for intramolecular hydrogen bonding to the hydroxyl groups. Nevertheless, in comparison with the pyridine derivative 1, the thiophene analogue is more effective in solvent inclusion, giving rise to successful isolation of at least six inclusion species, involving n-PrOH, n-BuOH, acetone, DMSO, 1,4-dioxane (2a) and pyrrolidine (2b) as guest solvents. This means that 2 is rather broad in solvent inclusion but actually does not come near to Ph (12 substantiated inclusions). For all that, 2 is superior to Ph in the inclusion of the linear medium-sized alcohols n-PrOH and n-BuOH. In this latter respect, 2 and Ph behave exactly opposite, whereas considering the failure to include the small alcohols MeOH and EtOH, they are just the same.
:1 and 1:2 inclusions with 1,4-dioxane (1a) and toluene (1b). Hence, it seems to be clear that 1 is only effective in inclusion formation with apolar solvents, and also one should think that the pyridine nitrogens are not involved in a specific host–guest interaction but are possibly used up for intramolecular hydrogen bonding to the hydroxyl groups. Nevertheless, in comparison with the pyridine derivative 1, the thiophene analogue is more effective in solvent inclusion, giving rise to successful isolation of at least six inclusion species, involving n-PrOH, n-BuOH, acetone, DMSO, 1,4-dioxane (2a) and pyrrolidine (2b) as guest solvents. This means that 2 is rather broad in solvent inclusion but actually does not come near to Ph (12 substantiated inclusions). For all that, 2 is superior to Ph in the inclusion of the linear medium-sized alcohols n-PrOH and n-BuOH. In this latter respect, 2 and Ph behave exactly opposite, whereas considering the failure to include the small alcohols MeOH and EtOH, they are just the same.
Table 1 Inclusion compounds formed from diol hosts 1 and 2 and from the literature known benzene analogue Ph
|
|
Ph
|
1
|
2
|
|
c—difficult to crystallize. |
| MeOH |
— |
— |
— |
| EtOH |
— |
— |
— |
|
n-PrOH |
— |
— |
1:2 |
|
n-BuOH |
— |
— |
1:2 |
| i-PrOH |
3:2 |
— |
— |
|
t-BuOH |
2:1 |
— |
— |
| Acetone |
2:1 |
— |
2:1 |
| Acetonitrile |
2:1 |
— |
— |
| Nitromethane |
2:1 |
— |
— |
| DMF |
1:2 |
— |
— |
| DMSO |
1:2 |
— |
2:1 |
| Et2NH |
— |
— |
— |
| Toluene |
— |
1:2 (1b) |
— |
| Pyridine |
1:2 |
— |
c
|
| THF |
2:1 |
— |
c
|
| 1,4-Dioxane |
1:2 |
1:1 (1a) |
1:1 (2a) |
| Pyrrolidine |
— |
— |
1:2 (2b) |
| Morpholine |
1:1 |
— |
c
|
| Piperidine |
1:2 |
— |
c
|
In order to gain more information about the possible reasons for the different behavior patterns of inclusion regarding the studied host compounds and including also the solvent-free species, X-ray structural analysis was undertaken.
X-ray structural study
Certainly, single crystals qualified for X-ray structural analysis have been obtained for the 1,4-dioxane (1a) and toluene (1b) inclusions of 1, but as far as 2 is concerned, suitable crystals could only be isolated from the inclusions with 1,4-dioxane (2a) and pyrrolidine (2b). Nevertheless, in some ways, a reasonable structural comparison is possible, in particular referring to the 1,4-dioxane inclusions 1a and 2a. Moreover, 1 and 3 (this latter compound exists in three different polymorphs) yielded suitable crystals for an X-ray study allowing further insight into structural circumstances. Crystallographic details and refinement parameters are summarized in Table 2.
Table 2 Crystallographic data and structure refinement details of the compounds studied
| Compound |
1
|
1a
|
1b
|
2a
|
2b
|
3A
|
3B
|
3C
|
|
P = (Fo2 + 2Fc2)/3.
|
| Empirical formula |
C28H22N4O2 |
C28H22N4O2·C4H8O2 |
C28H22N4O2·2C7H8 |
C24H18O2S4·C4H8O2 |
C24H18O2S4·2C4H9N |
C28H22N4O2 |
C28H22N4O2 |
C28H22N4O2 |
| Formula weight |
446.50 |
534.60 |
446.50 |
554.73 |
608.87 |
446.50 |
446.50 |
446.50 |
| Crystal system |
Monoclinic |
Triclinic |
Triclinic |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
Triclinic |
| Space group |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P21/c |
P21/c |
P |
P |
|
a (Å) |
8.1858(2) |
8.0741(2) |
8.1184(2) |
7.91030(10) |
10.8407(2) |
8.3478(18) |
8.2873(10) |
10.6246(3) |
|
b (Å) |
11.5886(2) |
8.8856(2) |
9.9587(3) |
8.5838(2) |
7.9198(2) |
8.0618(17) |
10.6627(12) |
12.9366(3) |
|
c (Å) |
12.1497(2) |
10.7962(3) |
10.9050(3) |
11.5231(2) |
17.3513(4) |
32.729(7) |
12.9725(18) |
16.5166(5) |
|
α (°) |
90.0 |
94.1790(10) |
106.597(2) |
109.7130(10) |
90.0 |
90.0 |
97.625(7) |
107.1770(10) |
|
β (°) |
108.4400(10) |
107.2500(10) |
108.7020(10) |
90.6290(10) |
96.9680(10) |
90.315(6) |
107.153(7) |
93.355(2) |
|
γ (°) |
90.0 |
108.9430(10) |
100.101(2) |
115.9310(10) |
90.0 |
90.0 |
93.365(9) |
97.5580(10) |
|
V (Å3) |
1093.37(4) |
687.28(3) |
764.91(4) |
650.70(2) |
1478.71(6) |
2202.6(8) |
1079.8(2) |
2138.65(10) |
|
Z
|
2 |
1 |
1 |
1 |
2 |
4 |
2 |
4 |
|
F(000) |
468 |
282 |
234 |
290 |
644 |
936 |
684 |
936 |
|
D
c (Mg m−3) |
1.356 |
1.292 |
0.969 |
1.416 |
1.367 |
1.346 |
1.373 |
1.387 |
|
μ (mm−1) |
0.088 |
0.087 |
0.063 |
0.399 |
0.355 |
0.087 |
0.089 |
0.090 |
| Data collection |
|
|
|
|
|
|
|
|
| Temperature (K) |
100(2) |
100(2) |
100(2) |
100(2) |
100(2) |
100(2) |
100(2) |
100(2) |
| No. of collected reflections |
26372 |
14885 |
16345 |
14288 |
16899 |
6041 |
17410 |
45269 |
| Within the θ limit (°) |
2.49–25.00 |
2.01–24.99 |
2.45–25.00 |
2.76–27.50 |
2.83–25.00 |
1.24–25.00 |
1.94–25.00 |
1.30–25.00 |
| Index ranges ±h, ±k, ±l |
−9/9, −13/13, −14/14 |
−9/9, −10/10, −12/12 |
−9/9, −11/11, −12/12 |
−10/10, −11/11, −14/14 |
−12/12, −8/9, −20/20 |
−5/9, −9/5, −23/38 |
−9/9, −12/12, −15/15 |
−12/12, −15/15, −19/19 |
| No. of unique reflections |
1925 |
2425 |
2694 |
2991 |
2593 |
3086 |
3799 |
7540 |
|
R
int
|
0.0232 |
0.0231 |
0.0248 |
0.0227 |
0.0216 |
0.0495 |
0.0282 |
0.0426 |
| Refinement calculations: full-matrix least-squares on all F2 values |
|
|
|
|
|
|
|
|
| Weighting expression wa |
[σ2(Fo2) + (0.0520P)2 + 0.6847P]−1 |
[σ2(Fo2) + (0.0429P)2 + 0.2391P]−1 |
[σ2(Fo2) + (0.0766P)2 + 0.1732P]−1 |
[σ2(Fo2) + (0.0805P)2 + 1.7941P]−1 |
[σ2(Fo2) + (0.0316P)2 + 0.9746P]−1 |
[σ2(Fo2) + (0.0467P)2]−1 |
[σ2(Fo2) + (0.0365P)2 + 0.4248P]−1 |
[σ2(Fo2) + (0.0633P)2]−1 |
| No. of refined parameters |
155 |
182 |
155 |
159 |
191 |
309 |
309 |
617 |
| No. of F values used [I > 2σ(I)] |
1807 |
2179 |
2453 |
2735 |
2457 |
1771 |
3199 |
3101 |
| Final R indices |
|
|
|
|
|
|
|
|
|
R (=∑|ΔF|/∑|Fo|) |
0.0409 |
0.0333 |
0.0421 |
0.0577 |
0.0268 |
0.0556 |
0.0356 |
0.0378 |
| wR on F2 |
0.1070 |
0.0854 |
0.1199 |
0.1607 |
0.0683 |
0.1037 |
0.0843 |
0.1076 |
|
S (goodness of fit on F2) |
1.086 |
1.081 |
1.097 |
1.091 |
1.048 |
0.938 |
1.060 |
0.939 |
| Final Δρmax/Δρmin (e Å−3) |
0.318/−0.287 |
0.237/−0.212 |
0.219/−0.227 |
1.791/−0.893 |
0.368/−0.213 |
0.191/−0.219 |
0.275/−0.194 |
0.206/−0.194 |
Crystal structures involving pyridino diol 1
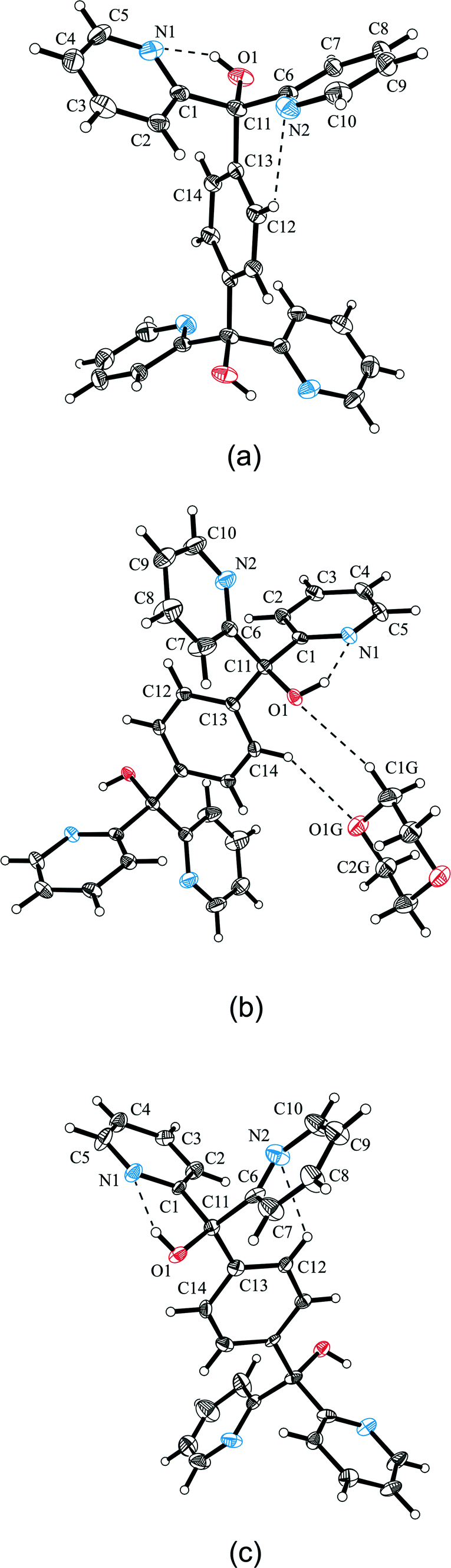
The solvent-free structure of pyridino diol 1 crystallizes as colorless prisms in the monoclinic space group P21/c from DMSO, DMF, acetone, pyridine and CHCl3 containing half a molecule in the asymmetric part of the unit cell. By way of contrast, the solvent inclusions of 1 with 1,4-dioxane 1a (1:1) and toluene 1b (1:2) crystallize in the triclinic space group P. The asymmetric part of the unit cell of 1a contains half a host and half a guest molecule. Due to the heavy disorder of toluene in 1b, the appropriate electron density was removed by the SQUEEZE method of the PLATON program,30 and consequently, the structure was refined without a solvent. Therefore, the asymmetric part of the unit cell in Fig. 2c shows only half a host entity. The molecular structures of all compounds connected with 1 are illustrated in Fig. 2. The respective conformations of the diol molecules in 1, 1a and 1b can be compared by the torsion angles of the pyridine and benzene rings with regard to the hydroxyl groups whereby only slight differences are recognizable (Table 3). This can be explained by the stabilizing intramolecular O–H⋯N and C–H⋯N interactions specified together with the intermolecular interactions in Table 4.
 |
| | Fig. 2 Molecular structures of solvent-free compound 1 (a) and host–guest inclusion compounds 1a (b) and 1b (c) shown as ellipsoid plots with 50% probability level. | |
Table 3 Torsion angles in the crystal structures of 1, 1a, 1b, 2a, and 2b
| Torsion angles (°) |
1
|
1a
|
1b
|
|
τ
1 (N1–C1–C11–O1) |
−6.24(2) |
−14.3(1) |
−22.9(1) |
|
τ
2 (N2–C6–C11–O1) |
163.1(1) |
136.9(1) |
160.3(1) |
|
τ
3 (C14–C13–C11–O1) |
−31.7(2) |
−39.7(1) |
−37.7(1) |
|
|
|
2a
|
2b
|
|
τ
1 (S1–C1–C9–O1) |
|
−29.3(1) |
−21.2(3) |
|
τ
2 (S2–C5–C9–O1) |
|
−27.8(1) |
−32.6(3) |
|
τ
3 (C12–C10–C9–O1) |
|
−58.7(2) |
−63.1(2) |
Table 4 Non-covalent interactions in the crystal structures of 1, 1a and 1b
| Atoms involved |
Symmetry |
Distance (Å) |
Angle (°) |
|
Cg3 is defined as the geometric centre of C12–C14 and C12′–C14′.
Cg1 is defined as the geometric centre of C1–C5 and N1.
|
|
|
|
D⋯A |
H⋯A |
D–H⋯A |
|
1
|
|
|
|
|
| O1–H1⋯N1 |
x, y, z |
2.577(2) |
2.03 |
123.9 |
| C12–H12⋯N2 |
x, y, z |
3.130(2) |
2.48 |
127.3 |
| C2–H2⋯O1 |
−x, 1/2 + y, 1/2 − z |
3.221(2) |
2.55 |
129.0 |
| C9–H9⋯O1 |
−1 − x, 1/2 + y, 1/2 − z |
3.457(2) |
2.63 |
148.2 |
| C8–H8⋯Cg3a |
x, y, z |
3.564(2) |
2.68 |
160.0 |
|
1a
|
|
|
|
|
| O1–H1⋯N1 |
x, y, z |
2.605(1) |
2.13 |
115.6 |
| O1–H1⋯N1 |
−x, 1 − y, 2 − z |
2.984(1) |
2.37 |
130.7 |
| C4–H4⋯O1 |
−1 + x, y, z |
3.342(2) |
2.52 |
144.9 |
| C5–H5⋯O1 |
−x, 1 − y, 2 − z |
3.098(2) |
2.56 |
116.1 |
| C8–H8⋯O1G |
x, −1 + y, z |
3.305(2) |
2.47 |
146.3 |
| C14–H14⋯O1G |
x, y, z |
3.468(2) |
2.62 |
149.3 |
| C1G–H1G1⋯O1 |
x, y, z |
3.570(2) |
2.61 |
162.2 |
| C2G–H2G2⋯N2 |
1 + x, 1 + y, z |
3.382(2) |
2.63 |
132.4 |
| C3–H3⋯Cg3a |
−1 + x, y, z |
3.790(1) |
2.96 |
146.7 |
| C9–H9⋯Cg1b |
x, −1 + y, z |
3.636(1) |
2.77 |
151.6 |
|
1b
|
|
|
|
|
| O1–H1⋯N1 |
x, y, z |
2.615(1) |
2.14 |
115.8 |
| C12–H12⋯N2 |
x, y, z |
3.187(2) |
2.55 |
124.9 |
| O1–H1⋯N1 |
1 − x, 2 − y, 1 − z |
2.994(1) |
2.37 |
131.6 |
| C4–H4⋯O1 |
−1 + x, y, z |
3.347(1) |
2.44 |
160.7 |
| C5–H5⋯O1 |
1 − x, 2 − y, 1 − z |
3.096(2) |
2.53 |
118.6 |
| C10–H10⋯N2 |
−x, 1 − y, −z |
3.527(2) |
2.70 |
146.2 |
| C3–H3⋯Cg3a |
−1 + x, y, z |
3.779(1) |
2.97 |
143.3 |
In the solvent-free structure of 1, the diol molecules are arranged in strands in the direction of the crystallographic a-axis connected by the C–H⋯π interaction31 C8–H8⋯Cg3 [d(C8⋯Cg3) = 3.564(2) Å] as shown in Fig. 3a. Along the c-axis, the host strands are linked by weak C–H⋯O hydrogen bonds32,33 C2–H2⋯O1 [d(C2⋯O1) = 3.221(2) Å] and C9–H9⋯O1 [d(C9⋯O1) = 3.457(2) Å], while in the b-direction, only π⋯π contacts between pyridine rings exist.34 The packing structure (Fig. 3b) offers no solvent accessible void, and consequently, a packing index (KPI)35 of 70.1% was determined.
 |
| | Fig. 3 (a) Molecular strand along the a-axis containing C–H⋯π contacts and (b) packing structure of solvent-free compound 1. Non-relevant H atoms were omitted for clarity. | |
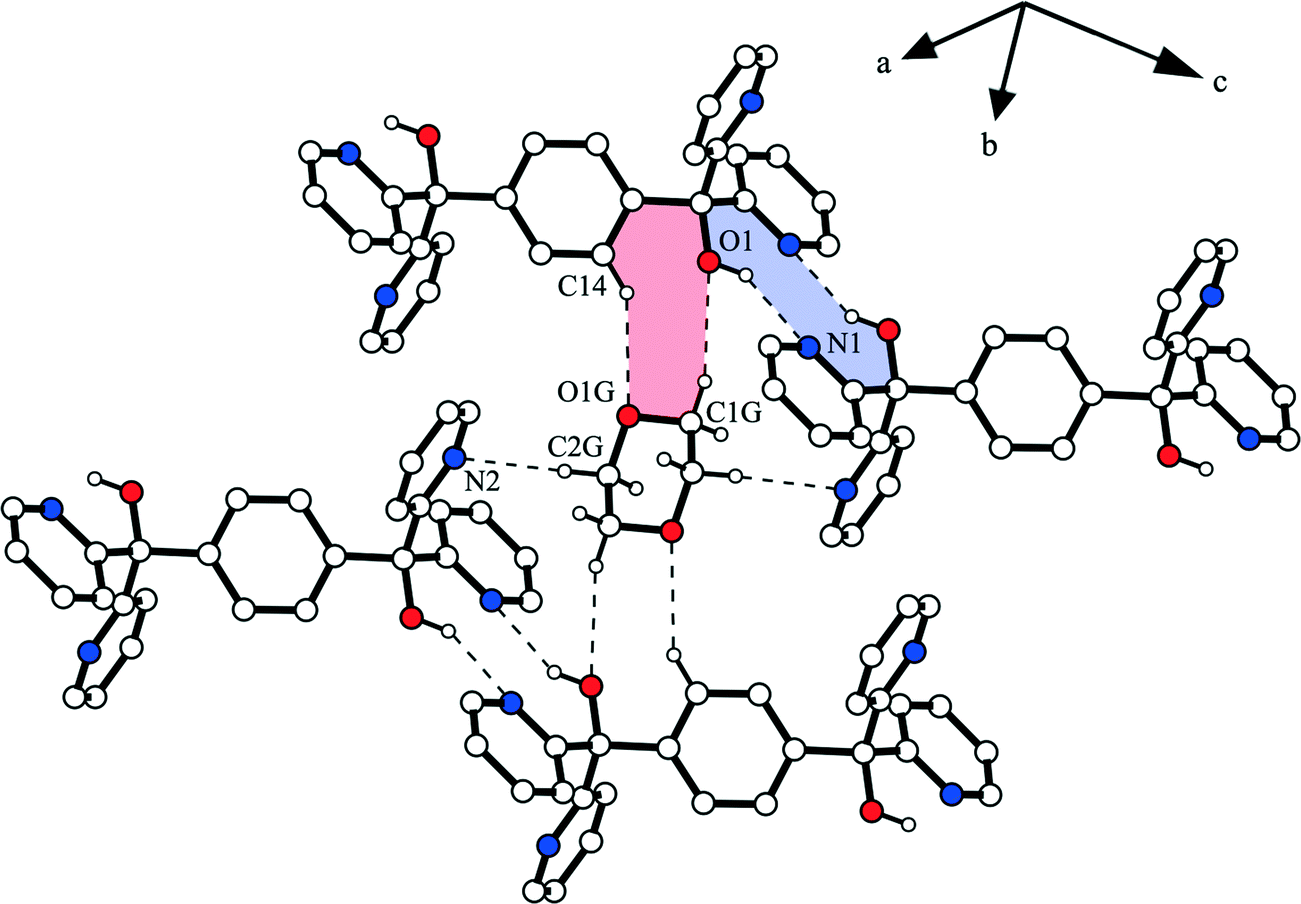
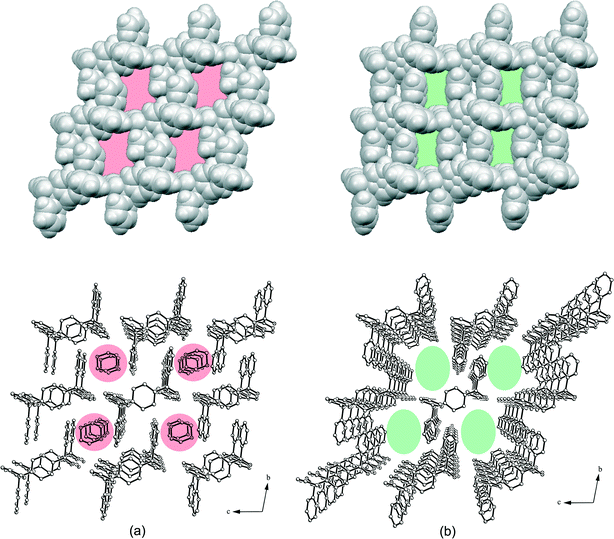
Inclusion compounds 1a and 1b consist of chain-like connected aggregates of host molecules that are stabilized along the c-axis by a hydrogen bond ring motif with the Etter/Bernstein graph set36,37 R22(10) highlighted for 1a in Fig. 4. Here, strong O–H⋯N interactions38 O1–H1⋯N1 [d(O1⋯N1) = 2.984(1) Å (1a) and d(O1⋯N1) = 2.994(1) Å (1b)] are involved. These chains are linked in the a-direction by the C–H⋯O hydrogen bond C4–H4⋯O1 [d(C4⋯O1) = 3.342(2) Å (1a) and d(C4⋯O1) = 3.347(1) Å (1b)], while in addition C–H⋯π contacts C3–H3⋯Cg3 [d(C3⋯Cg3) = 3.790(1) Å (1a) and d(C3⋯Cg5) = 3.775(2) Å (1b)] occur between methine groups of the pyridines and the benzene rings. The chains of 1a are connected by means of C–H⋯π interactions [d(C9⋯Cg1) = 3.636(1) Å] along the b-axis, whereas the stabilization of 1b is achieved by a hydrogen bond ring motif with the graph set R22(6) containing C–H⋯N contacts32 [d(C10⋯N2) = 3.527(2) Å]. Thus, solvent channels are formed along the a-axis containing 1,4-dioxane (1a, ca. 4.5 × 6.8 Å2) and toluene (1b, ca. 6.1 × 8.4 Å2), respectively (Fig. 5). The 1,4-dioxane molecules in 1a are fixed in the channels via weak C–H⋯O contacts [d(C1G⋯O1) = 3.570(2) Å, d(C8⋯O1G) = 3.305(2) Å and d(C14⋯O1G) = 3.468(2) Å] in which the guest oxygen O1G is inversely bifurcated and the hydroxyl oxygen O1 is inversely trifurcated. As a result of the dioxane connection, a further hydrogen bond ring motif with the graph set R22(8) is formed (Fig. 4). In addition, C–H⋯N interactions [d(C2G⋯N2) = 3.382(2) Å] exist between the methylene group of the dioxane and the pyridine nitrogen.
 |
| | Fig. 4 Interactions of the 1,4-dioxane molecule in the structure of 1a. The hydrogen bond ring motifs R22(8) and R22(10) are highlighted. Non-relevant H atoms are omitted for clarity. | |
 |
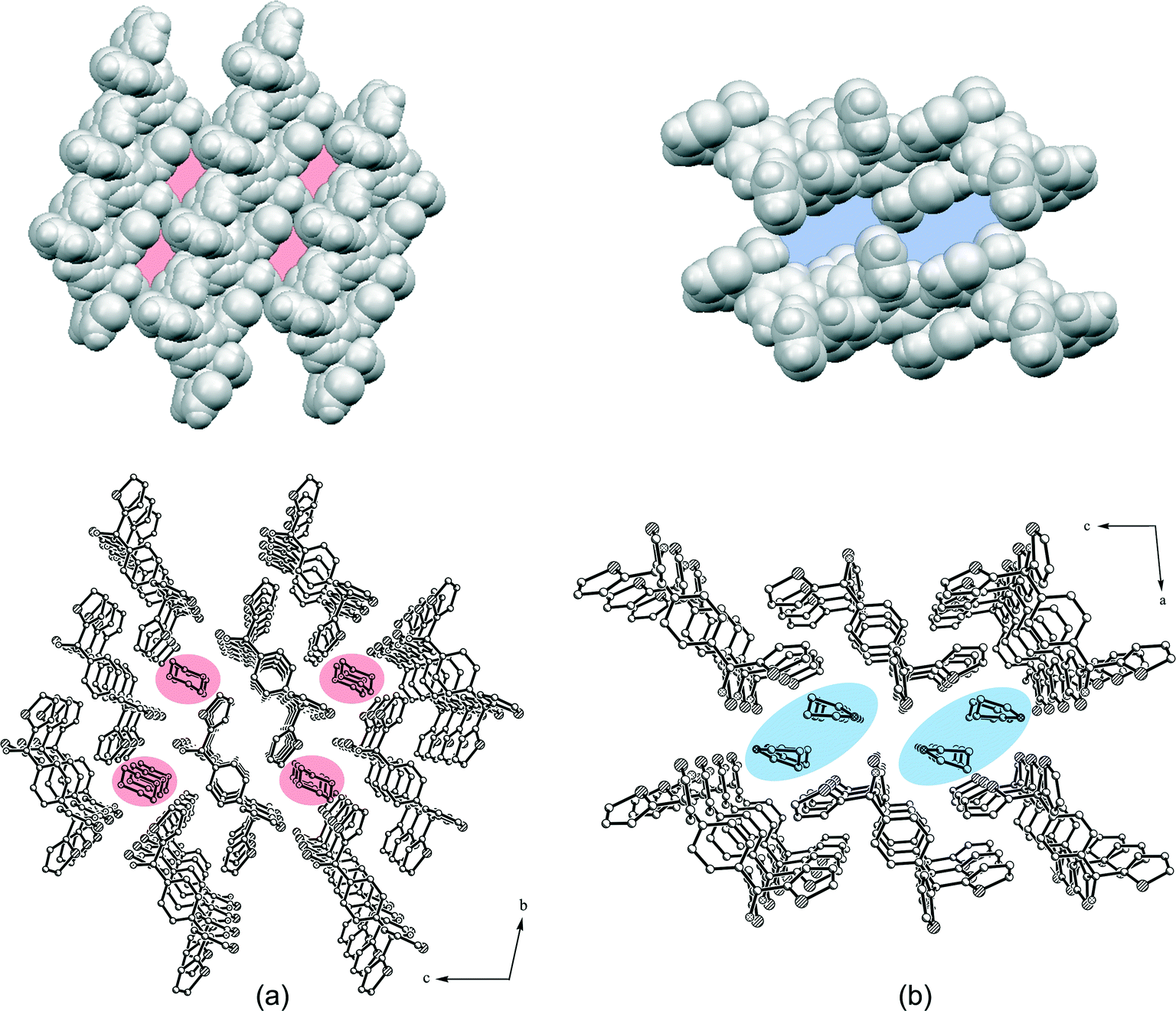
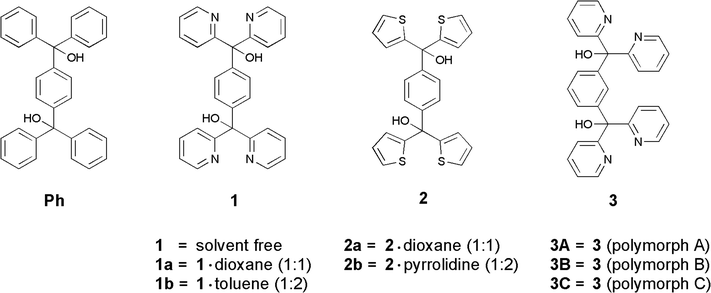
| | Fig. 5 Packing structure and van der Waals models (without guests) of the 1:1 1,4-dioxane inclusion compound 1a (a) and the 1:2 toluene inclusion compound 1b (b). The solvent channels along the a-axis are highlighted. All H atoms of the packing structures were omitted for clarity. | |
The toluene guest solvent in the structure of 1b is probably connected only by very weak interactions. Therefore, the high thermal motion prevents the refinement of the solvent molecules. Accordingly, the corresponding electron density was removed and the structure was refined without toluene. A solvent accessible void of 285.4 Å3, which equates to 37.3% of the unit cell, gives rise to a 1:2 host:guest stoichiometry, and the structure features a packing index (KPI) of 50.2% without a guest. In order to compare 1b with the dioxane inclusion compound 1a, the solvent was also removed from 1a yielding a solvent accessible void of 184.5 Å3, which equates to 26.8% of the unit cell (1:1 stoichiometry). Moreover, 1a features packing indices of 55.8% without dioxane and 67.7% with dioxane.
Crystal structures involving thiopheno diol compound 2
Thiopheno diol 2 crystallizes from 1,4-dioxane as a 1:1 host–guest inclusion (2a) in the triclinic space group P, while crystallization from pyrrolidine results in the formation of a 1:2 solvent inclusion (2b) in the monoclinic space group P21/c. The asymmetric parts of the unit cells contain half a dioxane molecule (2a) and half a host molecule as well as a pyrrolidine (2b), respectively, as shown in Fig. 6. A similarity in the molecular conformations of 2a and 2b is obvious regarding the torsion angles of the thiophene and benzene rings with respect to the hydroxyl groups (Table 3). Nevertheless, the packing arrangements of both structures markedly differ from each other (Fig. 7). Non-covalent interactions of 2a and 2b are summarized in Table 5.
 |
| | Fig. 6 Molecular structures of (a) 1,4-dioxane (2a) and (b) pyrrolidine (2b) host–guest inclusion compounds of 2 shown as ellipsoid plots with 50% probability level. | |
 |
| | Fig. 7 (a) Connection of the 1,4-dioxane guest in inclusion compound 2a by O–H⋯O and C–H⋯O hydrogen bonds as well as C–H⋯π interactions. The hydrogen bond ring motif R12(7) is highlighted. (b) Connection mode of the host molecules of 2b by the highlighted hydrogen bond ring motif R44(8) involving two pyrrolidine molecules. Non-relevant H atoms are omitted for clarity. | |
Table 5 Intermolecular interactions in the crystal structures of 2a and 2b
| Atoms involved |
Symmetry |
Distance (Å) |
Angle (°) |
|
Cg1 is defined as the geometric centre of C1–C4 and S1.
Cg5 is defined as the geometric centre of C2–C3.
Cg2 is defined as the geometric centre of C5–C8 and S2.
Cg4 is defined as the geometric centre of C10–C11.
Cg3 is defined as the geometric centre of C10–C12 and C10′–C12′.
|
|
|
|
D⋯A |
H⋯A |
D–H⋯A |
|
2a
|
|
|
|
|
| S2⋯O1 |
2 − x, 2 − y, 1 − z |
3.319(3) |
|
|
| S2⋯S2 |
2 − x, 2 − y, 1 − z |
3.533(3) |
|
|
| O1–H1⋯O1G |
x, y, z |
2.810(2) |
1.98 |
168.5 |
| C12–H12⋯O1G |
x, y, z |
3.483(2) |
2.70 |
140.6 |
| C1G–H1G1⋯Cg1a |
2 − x, 2 − y, 1 − z |
3.538(2) |
2.88 |
124.9 |
| C2G–H2G2⋯Cg5b |
−1 + x, −1 + y, z |
3.511(2) |
2.75 |
133.6 |
| C4–H4⋯Cg2c |
x, 1 + y, z |
3.553(2) |
2.79 |
137.5 |
| C7–H7⋯Cg4d |
1 + x, y, z |
3.665(2) |
2.73 |
168.3 |
|
2b
|
|
|
|
|
| O1–H1⋯N1G |
x, y, z |
2.685(2) |
1.88 |
161.0 |
| N1G–H1G⋯O1 |
1 − x, 1 − y, 1 − z |
3.085(2) |
2.23(2) |
171(2) |
| C4–H4⋯O1 |
x, −1 − y, z |
3.460(2) |
2.71 |
136.5 |
| C12–H12⋯N1G |
x, y, z |
3.464(2) |
2.70 |
137.9 |
| C2G–H2G1⋯S2 |
x, 3/2 − y, 1/2 + z |
3.729(2) |
2.96 |
134.8 |
| C3–H3⋯Cg3e |
x, −1 + y, z |
3.814(2) |
2.87 |
176.4 |
| C3G–H3G1⋯Cg2c |
x, 1/2 − y, 1/2 + z |
3.714(2) |
2.86 |
144.4 |
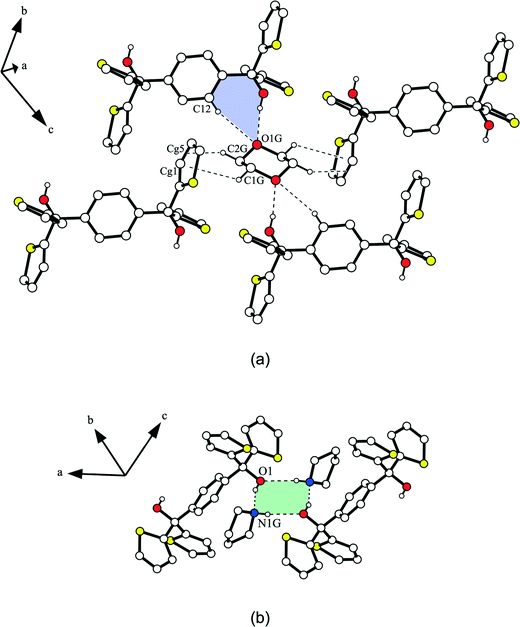
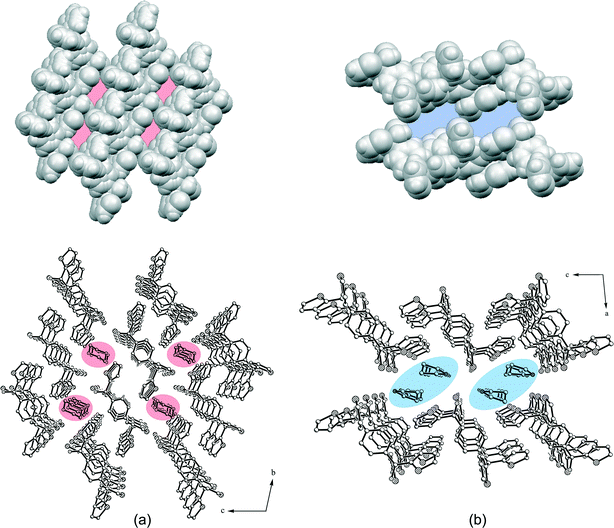
The host molecules of 2a are chain-like connected in the [111]-direction by S⋯O39 [d(S2⋯O1) = 3.319(3) Å] and S⋯S40 [d(S2⋯S2) = 3.533(3) Å] contacts.41,42 Along the a-axis, C–H⋯π interactions [d(C7⋯Cg4) = 3.665(2) Å] stabilize the structure, while in the b-direction the host molecules form strands linked by C–H⋯π contacts [d(C4⋯Cg2) = 3.553(2) Å]. This gives rise to the formation of solvent channels (ca. 2.8 × 4.2 Å2) along the crystallographic a-axis (Fig. 8a) in which the dioxane is fixed by strong O–H⋯O [d(O1⋯O1G) = 2.810(2) Å] and weak C–H⋯O [d(C12⋯O1G) = 3.483(2) Å] hydrogen bonds with the dioxane oxygen O1G being in an inverse bifurcated connection (Fig. 7a). Furthermore, C–H⋯π interactions of the dioxane methylene groups C1G–H1G1 [d(C1G⋯Cg1) = 3.538(2) Å] and C2G–H2G2 [d(C2G⋯Cg5) = 3.511(2) Å] with a thiophene ring exist in the structure. The packing index of 2a is 69.7%, while the structure without a guest features a solvent accessible void of 155.3 Å3 which equates to 23.9% of the unit cell. The corresponding solvent-free packing index was determined to be 57.5%.
 |
| | Fig. 8 Packing structure and van der Waals models (without guests) of the 1:1 dioxane inclusion compound 2a (a) and the 1:2 pyrrolidine inclusion compound 2b (b). The solvent channels along the a-axis are highlighted. All H atoms of the packing structures were omitted for clarity. | |
As contrasted with 2a, the host molecules of 2b are connected in strands along the crystallographic b-axis by weak C–H⋯O hydrogen bonds [d(C4⋯O1) = 3.460(2) Å] and C–H⋯π contacts [d(C3⋯Cg3) = 3.814(2) Å], while all further interactions exclusively involve host and guest molecules. Thus, a hydrogen bond ring motif with the graph set R44(8) including the OH groups of two host molecules bridged by the NH functions of two pyrrolidine molecules is observed (Fig. 7b). In addition, C–H⋯N contacts of the benzene methine group C12–H12 and the pyrrolidine nitrogen N1G [d(C12⋯N1G) = 3.464(2) Å] as well as C–H⋯π interactions of the pyrrolidine methylene group C3G–H3G1 and the thiophene ring Cg2 [d(C3G⋯Cg2) = 3.714(2) Å] stabilize the structure. Moreover, a methylene group of the pyrrolidine (C2G–H2G1) forms a C–H⋯S contact32,43 with the thiophene sulfur S2 [d(C2G⋯S2) = 3.729(2) Å]. Hence, in the packing structure, solvent channels (ca. 4.2 × 8.7 Å2) along the a-axis contain pyrrolidine guest molecules (Fig. 8b). A packing index (KPI) of 70.4% was determined for this structure. Removal of the guest from the structure offers a solvent accessible void of 472.5 Å3 which equates to 32.0% of the unit cell and the corresponding packing index amounts to 50.4%.
Crystal structures of the polymorphs of compound 3
According to our findings, compound 3 exclusively crystallizes solvent-free. But under different conditions, three polymorphs 3A–3C were obtained. Among them, polymorph 3A seems to be a very special case since it could be isolated only once, as a product batch of the synthesis of 3 after column chromatography, elution of the compound with ethyl acetate evaporation of the solvent and stirring with acetone yielded crystals of 3A in the monoclinic space group P21/c. Remarkably, we were unable to reproduce this result. By way of contrast, crystallization with cooling off and slow evaporation of 3 from solutions of EtOAc, CHCl3, pyridine, DMSO, DMF, acetone and n-BuOH, respectively, gave crystals of polymorph 3B in the triclinic space group P, while polymorph 3C crystallized from 1,4-dioxane in the space group as before.
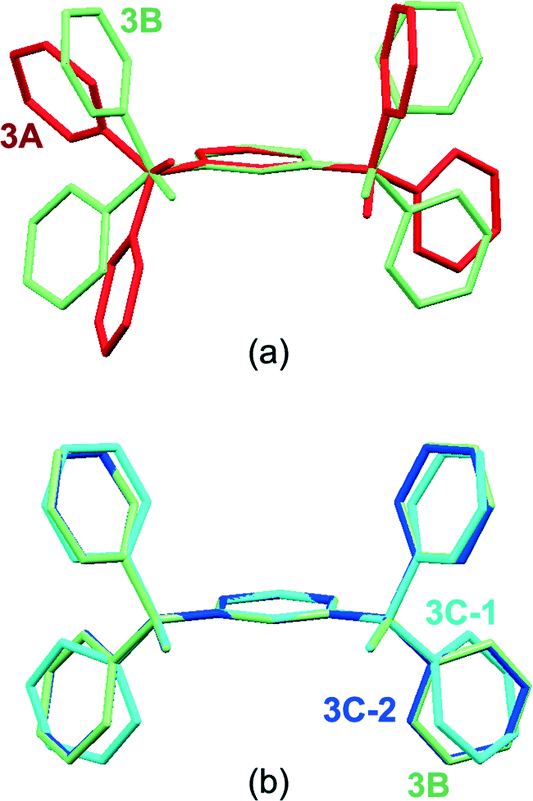
In the cases of 3A and 3B, the asymmetric part of the unit cell contains one diol molecule, whereas in 3C two independent diol molecules differing in the conformation and being also different from 3A are included (Fig. 9). This becomes apparent by the overlay of the central benzene unit and the tertiary sp3 carbon atoms in Fig. 10. While the oxygen atoms of 3B and 3C are nearly located within the benzene plane, the pyridine rings point away from the benzene plane (Fig. 10b). Moreover, the conformation of 3B is almost identical to that of molecule 2 of 3C, whereas molecule 1 of 3C features slightly twisted pyridine rings of which two pyridine nitrogens are in opposite orientation compared with molecule 2 of 3C. By way of contrast, the oxygen atoms of 3A are arranged out of the benzene plane in opposite directions, while two pyridine rings are nearly aligned to the benzene plane and the other two rings are almost vertical (Fig. 10a).
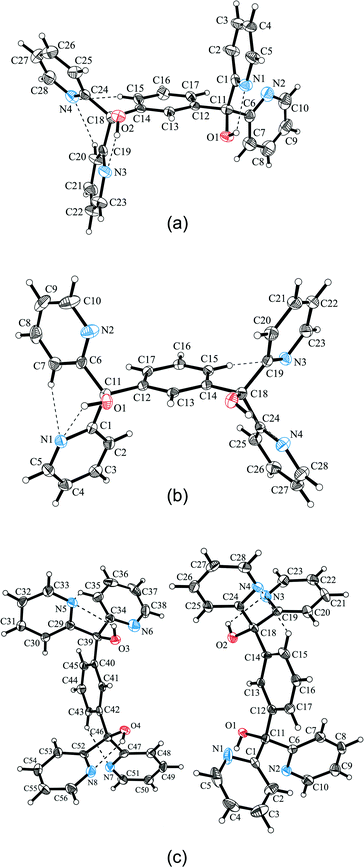 |
| | Fig. 9 Molecular structures of polymorphs 3A (a), 3B (b) and 3C (c) shown as ellipsoid plots with 50% probability level. | |
 |
| | Fig. 10 Overlay of the molecular conformations: (a) 3A and 3B, and (b) 3B and 3C (molecules 1 and 2). | |
A more detailed conformational comparison involving the torsion angles of the aromatic units with reference to the hydroxyl groups shows that corresponding values of the pyridine rings (−167.4° to 161.9°) are in a much broader range than the values of the benzene rings (−15.8° to 39.3°) (Table 6). Furthermore, the high conformational similarity between 3B and molecule 2 of 3C is confirmed by the torsion angles that are almost identical. Actually, the molecular conformations of all three polymorphs are stabilized by intramolecular O–H⋯N and C–H⋯N hydrogen bonding contacts including pyridine nitrogens and hydroxyl functions or benzene methine groups, respectively. The non-covalent interactions of the polymorphs of 3 are summarized in Table 7.
Table 6 Torsion angles of the molecular conformations of 3A, 3B and 3C
| Torsion angle (°) |
3A
|
3B
|
3C (molecule 1) |
3C (molecule 2) |
|
τ
1 (N1–C1–C11–O1) |
9.3(4) |
51.6(2) |
−53.8(4) |
51.6(4) (N5–C29–C39–O3) |
|
τ
2 (N2–C6–C11–O1) |
−139.3(3) |
53.8(2) |
−52.1(4) |
53.5(4) (N6–C34–C39–O3) |
|
τ
3 (C13–C12–C11–O1) |
39.3(4) |
14.9(2) |
−14.0(5) |
15.9(5) (C41–C40–C39–O3) |
|
τ
4 (N3–C19–C18–O2) |
−2.8(4) |
161.4(1) |
37.4(4) |
161.9(3) (N7–C47–C46–O4) |
|
τ
5 (N4–C24–C18–O2) |
−167.4(3) |
−37.4(2) |
−161.5(3) |
−37.1(4) (N8–C52–C46–O4) |
|
τ
6 (C13–C14–C18–O2) |
23.1(4) |
−15.1(2) |
14.9(4) |
−15.8(5) (C41–C42–C46–O4) |
Table 7 Intra- and intermolecular interactions of 3A, 3B and 3C
| Atoms involved |
Symmetry |
Distance (Å) |
Angle (°) |
|
Cg5 is defined as the geometric centre of C12–C17.
Cg4 is defined as the geometric centre of C24–C28 and N4.
Cg6 is defined as the geometric centre of C25–C26.
Cg11 is defined as the geometric centre of C20–C21.
Cg14 is defined as the geometric centre of C53–C54.
Cg12 is defined as the geometric centre of C27–C28.
|
|
|
|
D⋯A |
H⋯A |
D–H⋯A |
|
3A
|
|
|
|
|
| O1–H1⋯N1 |
x, y, z |
2.590(4) |
2.06 |
120.1 |
| O2–H2⋯N3 |
x, y, z |
2.505(4) |
1.94 |
123.7 |
| C15–H15⋯N4 |
x, y, z |
3.066(5) |
2.42 |
125.1 |
| C20–H20⋯N4 |
x, y, z |
3.008(5) |
2.58 |
107.9 |
| O1–H1⋯N1 |
1 − x, 1 − y, −z |
3.031(3) |
2.45 |
126.5 |
| C4–H4⋯O1 |
x, −1 + y, z |
3.399(4) |
2.59 |
142.6 |
| C5–H5⋯O1 |
1 − x, 1 − y, −z |
3.106(4) |
2.56 |
116.6 |
| C9–H9⋯O1 |
1 + x, y, z |
3.515(5) |
2.66 |
150.1 |
| C17–H17⋯O2 |
1 + x, y, z |
3.419(4) |
2.59 |
146.5 |
| C22–H22⋯O2 |
x, 1 + y, z |
3.307(5) |
2.44 |
152.3 |
| C3–H3⋯Cg5a |
x, −1 + y, z |
3.774(4) |
2.97 |
142.8 |
| C26–H26⋯Cg4b |
−x, −1/2 + y, 1/2 − z |
3.805(4) |
2.91 |
157.8 |
|
3B
|
|
|
|
|
| O1–H1⋯N1 |
x, y, z |
2.776(2) |
2.49 |
100.8 |
| O2–H2A⋯N4 |
x, y, z |
2.630(2) |
2.14 |
116.5 |
| C7–H7⋯N1 |
x, y, z |
3.122(2) |
2.66 |
110.1 |
| C15–H15⋯N3 |
x, y, z |
3.101(2) |
2.47 |
123.6 |
| O1–H1⋯N1 |
1 − x, −y, 1 − z |
2.822(2) |
2.07 |
148.6 |
| C5–H5⋯O1 |
1 − x, −y, 1 − z |
3.146(2) |
2.71 |
108.7 |
| C23–H23⋯N3 |
2 − x, 1 − y, −z |
3.448(2) |
2.70 |
136.0 |
| C27–H27⋯O2 |
1 + x, y, z |
3.466(2) |
2.65 |
143.9 |
| C28–H28⋯O1 |
2 − x, 1 − y, 1 − z |
3.426(2) |
2.48 |
173.6 |
| C5–H5⋯Cg6c |
2 − x, −y, 1 − z |
3.701(2) |
2.82 |
154.3 |
| C23–H23⋯Cg6c |
2 − x, 1 − y, −z |
3.613(2) |
2.87 |
136.2 |
|
3C
|
|
|
|
|
| O2–H2A⋯N3 |
x, y, z |
2.620(4) |
2.22 |
109.3 |
| C15–H15⋯N4 |
x, y, z |
3.089(5) |
2.47 |
122.8 |
| O3–H3A⋯N5 |
x, y, z |
2.766(4) |
2.34 |
111.6 |
| O4–H4A⋯N8 |
x, y, z |
2.622(4) |
2.11 |
118.6 |
| C35–H35⋯N5 |
x, y, z |
3.112(5) |
2.66 |
109.8 |
| C43–H43⋯N7 |
x, y, z |
3.077(5) |
2.44 |
124.2 |
| O1–H1⋯N5 |
1 + x, y, z |
2.817(4) |
2.02 |
156.9 |
| C10–H10⋯O3 |
1 + x, y, z |
3.129(5) |
2.69 |
108.8 |
| C22–H22⋯O4 |
1 − x, 1 − y, −z |
3.447(5) |
2.63 |
144.0 |
| C23–H23⋯O1 |
1 − x, 1 − y, −z |
3.420(5) |
2.48 |
172.3 |
| C28–H28⋯N4 |
1 − x, 2 − y, −z |
3.463(5) |
2.72 |
135.7 |
| O3–H3A⋯N2 |
−1 + x, y, z |
2.810(4) |
2.23 |
126.6 |
| C33–H33⋯O1 |
−1 + x, y, z |
3.143(5) |
2.71 |
108.7 |
| C51–H51⋯N7 |
1 − x, −y, 1 − z |
3.429(5) |
2.68 |
136.4 |
| C55–H55⋯O2 |
1 − x, 1 − y, 1 − z |
3.454(5) |
2.64 |
144.0 |
| C56–H56⋯O3 |
1 − x, 1 − y, 1 − z |
3.421(5) |
2.47 |
174.5 |
| C10–H10⋯Cg11d |
2 − x, 1 − y, −z |
3.691(4) |
2.81 |
154.4 |
| C28–H28⋯Cg11d |
1 − x, 2 − y, −z |
3.604(4) |
2.85 |
137.2 |
| C33–H33⋯Cg14e |
−x, 1 − y, 1 − z |
3.693(4) |
2.81 |
154.5 |
| C37–H37⋯Cg12f |
−x, 1 − y, −z |
3.671(4) |
2.83 |
148.7 |
| C51–H51⋯Cg14e |
1 − x, −y, 1 − z |
3.598(4) |
2.86 |
135.6 |
The packing structure of polymorph 3A consists of molecular dimers connected by a hydrogen bond ring motif with the graph set R22(10) including O–H⋯N contacts [d(O1⋯N1) = 3.031 Å], which is shown in Fig. 11a. Along the crystallographic a-axis, the dimers are arranged in strands stabilized by weak C–H⋯O contacts [d(C9⋯O1) = 3.515 Å and d(C17⋯O2) = 3.419 Å]. In the direction of the b-axis, the strands are also linked by C–H⋯O interactions [d(C4⋯O1) = 3.399 Å and d(C22⋯O2) = 3.307 Å] and in addition to that via C–H⋯π contacts [d(C3⋯Cg5) = 3.774 Å] with the oxygen atom O1 being in an inverse trifurcated connection and O2 in an inverse bifurcated connection. Along the c-axis, only C–H⋯π interactions [d(C26⋯Cg4) = 3.805 Å] exist between the strands.
 |
| | Fig. 11 Dimer strand in the packing structure of 3A (a) involving O–H⋯N and C–H⋯O interactions. The hydrogen bond ring motif R22(10) within the dimer is highlighted. Molecular chains of polymorphs 3B (b) and 3C (c) involving O–H⋯N and C–H⋯N interactions with hydrogen bond ring motifs R22(10) and R22(6) being specified. Non-relevant H atoms are omitted for clarity. | |
As contrasted with 3A, the packing structures of polymorphs 3B and 3C are arranged in a chain-like connection mode of the diol molecules (Fig. 11b and c). They are alternately linked by the already mentioned hydrogen bond ring motif R22(10) consisting of O–H⋯N interactions [d(O1⋯N1) = 2.822(2) Å (3B), d(O1⋯N5) = 2.817(4) Å and d(O3⋯N2) = 2.810(4) Å (3C)] and by a ring motif with the graph set R22(6) involving C–H⋯N contacts [d(C23⋯N3) = 3.448(2) Å (3B), d(C28⋯N4) = 3.463(5) Å and d(C51⋯N7) = 3.429(5) Å (3C)]. Due to the existence of two independent conformations in the structure of 3C, the diol chains feature an alternation of the set of independent conformations. In this case, molecules of the same conformation are connected by the above specified C–H⋯N contacts, while those possessing different conformations are linked by the O–H⋯N interactions. Superimposed molecular chains are stabilized in the direction of the a-axis by C–H⋯O [d(C17⋯O2) = 3.466 Å (3B) and d(C22⋯O4) = 3.447 Å (3C)] and C–H⋯π contacts [d(C5⋯Cg6) = 3.701 Å (3B) and d(C33⋯Cg14) = 3.693 Å (3C)] as well as by π⋯π interactions between pyridine rings. Along the [111]-direction, the chains are linked by weak C–H⋯O hydrogen bonds [d(C28⋯O1) = 3.426 Å (3B), d(C56⋯O3) = 3.421 Å and d(C23⋯O1) = 3.420 Å (3C)], while additional C–H⋯π contacts [d(C4⋯Cg13) = 3.641 Å and d(C37⋯Cg12) = 3.671 Å] exist in 3C.
Considering the thermal behavior of 3A–3C, it is obvious that polymorph 3A is the one that melts at the lowest temperature, i.e. from 187 to 189 °C, while 3B and 3C melt between 196 and 199 °C. More detailed investigation with reference to 3B (Sup. 1) and 3C (Sup. 2) has been performed by differential scanning calorimetry (DSC). Here, onset temperatures for melting of 198.6 °C (3B) and 197.6 °C (3C) as well as enthalpy values for the melting process of 46.8 kJ mol−1 (3B) and 46.0 kJ mol−1 (3C) have been determined. Actually a difference in melting temperature of 1 K regarding 3B and 3C is only small, nevertheless in a way significant since it correlates with the close relationship between the two crystal structures. Furthermore, one may conclude from the melting data that 3B is the most stable species in the series of the present polymorphs followed by 3C and 3A. To a certain extent, this is perhaps in line with the preferred result of the chemical preparation of 3, normally leading to 3B. The DSC diagrams are included in the ESI† as Sup. 1 and Sup. 2.
Sorption behavior
Apart from the static inclusion of guest molecules realized by crystallization, which has been discussed above, the dynamic sorption properties of host compounds are particularly interesting for application as mass sensitive sensor materials.28,44 Previous investigation along this line based on sorption behavior studies by QCM measurements45 has been carried out for other potential host compounds.46–50 Being deduced from it, a solvent-free host coating should be able to interact with gaseous guest molecules yielding a temporary inclusion compound. In order to investigate potential sorptive guest selectivity of the present compounds, different solvents representing varied polarity properties with protic as well as aprotic characteristics (Fig. 12) were used (n-hexane, CHCl3, THF, acetone, EtOH and Et2NH). One may expect that the rather limited inclusion ability of 1 and 2 derived from the above crystallization studies, but on the other hand indicating distinct selectivity, could also be reflected in the results of sorption measurements. On parallel testing of 1 and 2 towards different solvents, the thiophene containing host 2, in particular, behaves remarkably, clearly featuring a selective sorption of THF (84%) and Et2NH (108%, ca. 1:1 stoichiometry), while the other solvents were adsorbed only in amounts below 50%. Here, the sorption of Et2NH is a notable finding since formation of an inclusion compound with Et2NH was not observed under crystallization conditions, possibly due to the lack of a stable packing arrangement. But sorption of the amine from the gas phase seems to be definitely preferred probably owing to the formation of strong O–H⋯N and N–H⋯O interactions between the host hydroxyl and the guest amine functions. By way of contrast, the pyridine containing diol 1 shows a selective sorption of EtOH (73%), while the other solvents were adsorbed only below the index of 50%. An explanation for this remarkable behavior could be the formation of strong intramolecular hydrogen bonds (cf. crystal structure of unsolvated 1) preventing intermolecular H-donor properties of the host hydroxyl groups in hydrogen bonding to guests. Hence, 1 is only available as a H-acceptor in hydrogen bonding to active H-donor guest molecules such as EtOH. Following this point of view, protic Et2NH should also be a suitable candidate for binding to 1. However, contrary to expectation, Et2NH is not absorbed as compared to EtOH. Here, steric hindrance between the host and the rather bulky Et2NH molecule may account for this specific restriction. Moreover, it is very obvious from the diagram in Fig. 12 that both the aprotic solvents CHCl3 and n-hexane are distinctly less absorbed using 1 as well as 2, being in conformity with the above conception of host–guest hydrogen bonding.
 |
| | Fig. 12 Sorption behavior of the pyridino and thiopheno diol hosts 1 and 2 with respect to the used solvents (n-hexane, CHCl3, THF, acetone, EtOH, and Et2NH). | |
Comparative discussion and conclusion
Nuclear addition reactions of 2-pyridyllithium, 3-pyridyllithium or 2-thienyllithium to corresponding diacid dichlorides (1 and 3) or the corresponding diester (2) successfully gave new diol compounds 1–3 in moderate to high yields. Based on the heteroaromatic (pyridyl, thienyl) moieties substituting the phenyl residues of the model wheel-and-axle host molecule (Ph), they represent examples of a new structural modification of this particular host type featuring further potential receptor sites for hydrogen bonding (pyridyl) or other interaction modes emanating from the thienyl groups. However, X-ray structures involving pyridyl-containing 1 and 3 both in solvent-free and solvent-complexed forms show that the hydroxy groups are intramolecularly coordinated to pyridyl moieties which is restrictive to polar solvent interaction, while thiophene derivative 2 is not subjected to a corresponding behavior. This is clearly expressed by the number and variety of crystalline solvent inclusions obtained from 1 and 2 demonstrating that 1 has a much lower capability to include solvents than 2, being restraint only to the apolar solvent molecules 1,4-dioxane and toluene under the conditions of this study. Nevertheless, the host molecule 2, having a moderately broad inclusion behavior, is distinctly inferior in forming crystalline solvent inclusions to the prototype host compound Ph. Reflecting on a comparison between solvent inclusions of Ph and 2, there are a few rather close similarities, e.g. inclusion of both with acetone and toluene. But Ph failed in solvent inclusion with alcohols, while 2 proved its activity in this respect. Thus, on the one hand, both 1 and 2 can be seen as hosts inferior to Ph in a multitude of solvent inclusions, but it seems that they are superior to Ph in the selectivity of inclusion formation which, however, requires support from a more detailed study including competition of solvents. Remarkably, the 1,3-substituted pyridino derivative at the axle position (3) is found incapable of solvent inclusion pointing out that straightness of the axle moiety is the whole point of the host structure. Yet one has the impression that the packing of 3 is not a definite case allowing the formation of at least three polymorphs on crystallization from different solvents, unlike 1 that also crystallizes in a solvent-free form. By way of contrast, in the solvent inclusions of 1 and 2, the host–guest connectivity is caused by a variety of weak C–H⋯N and C–H⋯O contacts as well as strong O–H⋯N and O–H⋯O hydrogen bonds giving rise to noticeable ring motifs.
Furthermore, sorption experiments applying a quartz crystal microbalance coated with solid films of 1 and 2 proved efficient in the selective uptake of solvent vapors considering a variety of apolar to polar solvents that should offer a promising possibility for sensor development. However, the results of this study also make us aware that the vapor sorption behavior and solvent co-crystallization properties of the present hosts are only limitedly transferable. As a matter of fact, due to the different modes of inclusion formation, this difference appears evident.
Experimental
General remarks
The melting points were measured using a microscope heating stage (Thermovar, Reichert-Jung). IR spectra were recorded on a Nicolet FT-IR 510 spectrometer as KBr pellets (wavenumbers given in cm−1). 1H and 13C NMR spectra were obtained using a Bruker Avance 500 at 500.1 (1H) and 125.8 MHz (13C) with TMS as internal standard. Chemical shifts for proton and carbon resonances are given in ppm (δ). Mass spectra were recorded on a Hewlett Packard 5890 Series II/MS 5971 A. DSC measurements were executed using a Sensys TGA DSC (Setaram). TLC analysis was performed using aluminium sheets precoated with silica gel 60 F254 (Merck). Silica gel (63–100 μm, Merck) was used for column chromatography. Solvents were purified by standard procedures.
Syntheses of diols 1–3
1,4-Bis[di(pyrid-2-yl)hydroxymethyl]benzene (1).
To a mixture of 2-bromopyridine (1.96 g, 12.4 mmol) in 10 ml of dry THF, cooled to −78 °C under argon, n-BuLi (7.8 ml, 12.4 mmol, 1.6 M in n-hexane) was added slowly via a syringe. After stirring the solution for 15 min at −78 °C, terephthaloyl chloride (0.63 g, 3.1 mmol) dissolved in 10 ml of dry THF was added dropwise. The resulting mixture was stirred for 1 h at −78 °C, then for 3 h at room temperature and after that hydrolyzed with sat. NH4Cl solution. The mixture was extracted with CH2Cl2 (3 × 20 ml), the organic phase dried (Na2SO4) and the solvent evaporated. The obtained brown oil was treated under stirring for 20 min in 10 ml of acetone to give a precipitate which was separated and dried yielding compound 1 as a beige solid (0.51 g, 57%). Mp 229–232 °C (from acetone). NMR data: δH (500.1 MHz, CDCl3, TMS) 6.87 (2H, s, OH), 7.16 (4H, td, PyH5, JHH = 4.90, 0.90 Hz), 7.22 (4H, s, Ph), 7.65 (4H, td, PyH4, JHH = 7.65, 1.75 Hz), 7.76 (4H, d, PyH3, JHH = 7.95 Hz), 8.49 (4H, d, PyH6, JHH = 8.05 Hz). δC (125.8 MHz, CDCl3) 80.2 (C–OH), 122.2 (PyC5), 123.2 (PyC3), 127.5 (Ph), 136.4 (PyC4), 145.0 (Ph), 147.3 (PyC6), 163.0 (PyC2). IR data: νmax 3278m (OH, br), 3056w and 3003w (CHAr), 1739w, 1717w, 1584s and 1572m (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1464s (CN), 1432s and 1410s (OH), 1388m, 1299w, 1283w, 1249w, 1185w, 1150w, 1128w, 1100w, 1068m and 1052m (C–O), 1021m, 995m, 964w, 926w, 891w, 831w, 767s and 748s (CH), 707m, 672m (OH), 618w, 599w, 565w, 542w, 517w. MS data (ESI): calc. for C28H22N4O2 (446.17), found: 447.0 m/z [M + H]+. Anal. calc. for C28H22N4O2: C, 75.32; H, 4.97; N, 12.55, found: C, 75.49; H, 4.96; N, 12.30%.
C), 1464s (CN), 1432s and 1410s (OH), 1388m, 1299w, 1283w, 1249w, 1185w, 1150w, 1128w, 1100w, 1068m and 1052m (C–O), 1021m, 995m, 964w, 926w, 891w, 831w, 767s and 748s (CH), 707m, 672m (OH), 618w, 599w, 565w, 542w, 517w. MS data (ESI): calc. for C28H22N4O2 (446.17), found: 447.0 m/z [M + H]+. Anal. calc. for C28H22N4O2: C, 75.32; H, 4.97; N, 12.55, found: C, 75.49; H, 4.96; N, 12.30%.
1,4-Bis[di(thien-2-yl)hydroxymethyl]benzene (2).
The synthesis follows a described procedure.29 To a mixture of thiophene (1.75 g, 20.8 mmol) in 20 ml of dry THF, cooled to −30 °C under argon, n-BuLi (13.0 ml, 20.8 mmol, 1.6 M in n-hexane) was added slowly via a syringe. After stirring the solution for 15 min at −30 °C, dimethyl terephthalate (0.82 g, 4.2 mmol) dissolved in 20 ml of dry THF was added dropwise. The resulting mixture was allowed to warm to room temperature and after that hydrolyzed with sat. NH4Cl solution. The mixture was extracted with CH2Cl2 (3 × 30 ml), the organic phase dried (Na2SO4) and the solvent evaporated. The residue was purified by chromatography on silica gel (EtOAc/n-hexane, Rf: 0.78) to yield 2 as a white solid (1.42 g, 98%). Mp 155–158 °C (from acetone). NMR data: δH (500.1 MHz, CDCl3, TMS) 3.18 (2H, s, OH), 6.84 (4H, dd, ThH3, JHH = 3.52, 1.00 Hz), 6.94 (4H, m, ThH4), 7.27 (4H, dd, ThH5, JHH = 5.08, 1.03 Hz), 7.43 (4H, s, Ph). δC (125.8 MHz, CDCl3) 77.8 (C–OH), 125.8 (ThC3), 126.3 (ThC5), 126.6, 145.5 (Ph), 151.6 (ThC2). IR data: νmax 3528m (OH), 3104w and 3069w (CHAr), 2923w, 2854w, 1929w, 1806w, 1632w, 1502w, 1426m (OH), 1407m, 1353m (CC), 1312m, 1233m (CC), 1169w, 1131w, 1109w, 1084w, 1040w, 1021w, 989m, 875w, 853m, 837m, 818m, 796m, 780m, 701s (CH), 631w, 508w, 470w. MS data (ESI): calc. for C24H18O2S4 (466.02), found: 488.9 m/z [M + Na]+. Anal. calc. for C24H18O2S4: C, 61.77; H, 3.89; S, 27.48, found: C, 61.80; H, 3.91; S, 26.94%.
1,3-Bis[di(pyrid-2-yl)hydroxymethyl]benzene (3).
For the reaction, an analogous procedure to for 1 was applied although isophthaloyl chloride (0.63 g, 3.1 mmol) was used instead of terephthaloyl chloride. Purification of the compound was achieved as follows. The residue resulting from solvent removal was column chromatographed on silica gel (EtOAc, Rf: 0.59) and the evaporated eluate was stirred for 20 min in 10 ml of acetone. The solid was collected by filtration and dried to yield compound 3 as a beige powder (0.29 g, 33%). Mp 196–199 °C (from acetone). NMR data: δH (500.1 MHz, CDCl3, TMS) 6.80 (2H, s, OH), 7.13 (4H, td, PyH5, JHH = 7.35, 4.85, 1.15 Hz), 7.19–7.23 (4H, m, Ph), 7.60 (4H, td, PyH4, JHH = 7.40, 1.80 Hz), 7.67 (4H, dt, PyH3, JHH = 8.05, 1.10 Hz), 8.45 (4H, m, PyH6). δC (125.8 MHz, CDCl3) 80.4 (C–OH), 122.0 (PyC5), 123.1 (PyC3), 126.7, 127.6, 127.9 (Ph), 136.3 (PyC4), 145.6 (Ph), 147.2 (PyC6), 163.1 (PyC2). IR data: νmax 3354m (OH, br), 3056w and 3009w (CHAr), 2923w, 1584s and 1568m (CC), 1467s (CN), 1432s (OH), 1372m, 1296w, 1204w, 1150w, 1103w, 1049m (C–O), 1002m, 941m, 913w, 802m, 777m, 751s (CH), 713m, 681w, 656m (OH), 644w, 622w, 584w, 555w, 504w. MS data (ESI): calc. for C28H22N4O2 (446.17), found: 447.2 m/z [M + H]+. Anal. calc. for C28H22N4O2: C, 75.32; H, 4.97; N, 12.55, found: C, 75.04; H, 4.91; N, 12.25%.
Preparation of crystalline inclusion compounds
The host compound was dissolved by heating in a minimum amount of the respective guest solvent. After storage for 12 h at room temperature, the crystals which formed were collected and dried (1 h, 15 Torr, room temperature). The host:guest stoichiometric ratios were determined by 1H-NMR integration. The data for each compound are given in Table 1.
X-ray structure determination
Crystals of inclusion compounds 1a, 1b, 2a, and 2b suitable for X-ray diffraction were obtained by slow evaporation of solutions of 1 and 2 in the respective solvents (1,4-dioxane for 1a, toluene for 1b, 1,4-dioxane for 2a, and pyrrolidine for 2b). Crystals of the guest-free compound 1 were grown from DMSO and those of polymorphs 3A, 3B and 3C from acetone, CHCl3 and 1,4-dioxane, respectively. The intensity data were collected at 100 K using a Bruker Kappa diffractometer equipped with an APEX II CCD area detector and graphite-monochromatized Mo Kα radiation (λ = 0.71073 A) employing φ and ω scan modes. The data were corrected for Lorentz and polarization effects. Semi-empirical absorption correction was applied using the SADABS program.51 The SAINT program51 was used for the integration of the diffraction profiles. The crystal structures were solved by direct methods using SHELXS-97 (ref. 52) and refined by full-matrix least-squares refinement against F2 using SHELXL-97.52 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were positioned geometrically and allowed to ride on their parent atoms. Geometrical calculations were performed using PLATON and molecular graphics were generated using SHELXTL.52 One thiophene ring of the host molecule in structure 2b is twice disordered (sof = 0.80). The crystal structure of polymorph 3A was solved only with a data completeness of 80%. Unfortunately, we were unable to re-isolate this metastable polymorph under the mentioned conditions, putting us into a potential position to use a more complete data set.
Sorption measurements
For the sorption experiments, a quartz crystal microbalance consisting of two electronic quartzes (10 MHz) with gold electrodes (FOQ Piezo Technik, Germany) was used. The reference quartz is uncoated, while the other quartz is coated with the solid diol compound (1 and 2, respectively). The measurements were carried out at constant temperature (25 °C) and with a constant flow of synthetic air (10 L h−1). A multichannel frequency counter (HKR Sensor Systems, Munich, Germany) with a resolution of 1 Hz was used to measure the resonance frequencies of the quartzes which can be read by a computer using a serial interface. The coating of the quartz was realized by dipping in a 0.01 M solution of the respective diol compound in CHCl3. The change in frequency is proportional to the increase in quartz mass induced by the sorption of the added solvent vapor. This relation results from the Sauerbrey equation.53 In consideration of the molar mass of the used solvents, the percentage of the adsorbed solvent can be obtained.
Acknowledgements
Financial support from the European Union (European Regional Development Fund) and from the Ministry of Science and Art of Saxony (Cluster of Excellence “Structure Design of Novel High-Performance Materials via Atomic Design and Defect Engineering [ADDE]”) is gratefully acknowledged by FK and EW.
References
-
Comprehensive Supramolecular Chemistry, ed. D. D. MacNicol, F. Toda and R. Bishop, Elsevier, Oxford, U.K., 1996, vol. 6 Search PubMed.
-
T. Hertzsch, J. Hulliger, E. Weber and P. Sozzani, in Encyclopedia of Supramolecular Chemistry, ed. J. L. Atwood and J. W. Steed, CRC Press, Boca Raton, FL, 2004, pp. 996–1005 Search PubMed.
-
Organic Solid State Reactions, Topics in Current Chemistry, ed. F. Toda, Springer-Verlag, Berlin/Heidelberg, 2005, vol. 254 Search PubMed.
-
Metal-Organic Frameworks: Design and Application, ed. L. R. Gillivray, Wiley, Hoboken, NY, 2010 Search PubMed.
-
Inclusion Compounds, ed. J. L. Atwood, J. E. D. Davies and D. D. MacNicol, Oxford University Press, Oxford, U.K., 1991, vol. 4 Search PubMed.
-
E. Weber, in Kirk-Othmer Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz, Wiley, New York, 4th edn, 1995, vol. 14, pp. 122–154 Search PubMed.
-
A. Nangia, in Nanoporous Materials, ed. G. Q. Lu and X. S. Zhao, Series on Chemical Engineering, Imperial College Press, London, 2004, vol. 4, pp. 165–187 Search PubMed.
-
F. H. Herbstein, Crystalline Molecular Complexes and Compounds, Oxford University Press, Oxford, U.K., 2005, vol. 1, pp. 421–513 Search PubMed.
-
J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, New York, 2nd edn, 2009 Search PubMed.
-
Organic Crystal Engineering, ed. J. J. Vittal, M. Zaworotko and E. R. T. Tiekink, Wiley, New York, 2010 Search PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342 CrossRef CAS PubMed.
-
Making Crystals by Design: Methods, Techniques and Applications, ed. D. Braga and F. Creponi, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
- R. Bishop, Chem. Soc. Rev., 1996, 25, 311 RSC.
-
G. R. Desiraju, in Comprehensive Supramolecular Chemistry, ed. D. D. MacNicol, F. Toda and R. Bishop, Elsevier, Oxford, U.K., 1996, vol. 6, pp. 1–32 Search PubMed.
-
E. Weber, in Inclusion Compounds, ed. J. L. Atwood, J. E. D. Davies and D. D. MacNicol, Oxford University Press, Oxford, U.K., 1991, vol. 4, pp. 188–262 Search PubMed.
-
E. Weber and M. Czugler, in Molecular Inclusion and Molecular Recognition: Clathrates II, ed. E. Weber, Topics in Current Chemistry, Springer-Verlag, Berlin/Heidelberg, 1988, vol. 149, pp. 45–135 Search PubMed.
-
F. Toda, in Comprehensive Supramolecular Chemistry, ed. D. D. MacNicol, F. Toda and R. Bishop, Elsevier, U.K., 1996, vol. 6, pp. 465–516 Search PubMed.
- H. Hart, L.-T. W. Lin and D. L. Ward, J. Am. Chem. Soc., 1984, 106, 4043 CrossRef CAS.
- F. Toda, A. Kai, R. Toyotaka, W.-H. Yip and T. C. W. Mak, Chem. Lett., 1989, 1921 CrossRef CAS.
- E. Weber, K. Skobridis, A. Wierig and L. R. Nassimbeni, J. Chem. Soc., Perkin Trans. 2, 1992, 2123 RSC.
-
E. Weber, in Comprehensive Supramolecular Chemistry, ed. D. D. MacNicol, F. Toda and R. Bishop, Elsevier, U.K., 1996, vol. 6, pp. 535–592 Search PubMed.
- Y. Mazaki, N. Hayashi and K. Kobayashi, J. Chem. Soc., Chem. Commun., 1992, 1381 RSC.
- G. R. Newkome, J. D. Sauer and M. L. Erbland, J. Chem. Soc., Chem. Commun., 1975, 885 RSC.
- C. Bachmann, M. Guttentag, B. Spingler and R. Alberto, Inorg. Chem., 2013, 52, 6055 CrossRef CAS PubMed.
- D. A. Kurmaev, N. A. Kolosov, S. C. Gagieva, A. O. Borissova, V. A. Tuskaev, N. M. Bravaya and B. M. Bulychev, Inorg. Chim. Acta, 2013, 396, 136 CrossRef CAS.
- C. W. Thornber, Chem. Soc. Rev., 1979, 8, 563 RSC.
-
N. Brown, Bioisosters in Medicinal Chemistry, Wiley-VCH, Weinheim, 2012 Search PubMed.
-
Piezoelectric
Sensors, ed. A. J. C. Steinem and A. Janshoff, Springer-Verlag, Heidelberg, 2006 Search PubMed.
- H. Kurata, M. Inase and M. Oda, Chem. Lett., 1999, 519 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757 RSC.
-
G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond. In Structural Chemistry and Biology, Oxford University Press, New York, 1999 Search PubMed.
- D. Eißmann, F. Katzsch and E. Weber, Struct. Chem., 2012, 23, 1131 CrossRef.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
-
A. I. Kitaigorodskii, Molecular Crystals and Molecules, Academic Press, New York, 1973 Search PubMed.
- M. C. Etter, J. Phys. Chem., 1991, 95, 4601 CrossRef CAS.
- J. Bernstein, R. E. Davies, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
-
G. Gilli and P. Gilli, The Nature of the Hydrogen Bond, Oxford University Press, Oxford, 2009 Search PubMed.
- A. Kucsman, I. Kapovits, M. Czugler, L. Parkanyi and A. Kalman, J. Mol. Struct., 1989, 198, 339 CrossRef CAS.
- R. Gleiter, D. B. Werz and B. J. Rausch, Chem. – Eur. J., 2003, 9, 2676 CrossRef CAS PubMed.
- I.-L. Long, P. Kögerler and L. Cronin, Angew. Chem., Int. Ed., 2004, 43, 1817 CrossRef PubMed.
- M. Mascal, Contemp. Org. Synth., 1994, 1, 31 RSC.
- J. J. Novoa, M. C. Rovira, C. Rovira, J. Veciana and J. Tarres, Adv. Mater., 1995, 7, 233 CrossRef CAS.
-
Piezoelectric Transducers and Applications, ed. A. Arnau, Springer-Verlag, Heidelberg, 2004 Search PubMed.
- J. W. Grate, Chem. Rev., 2000, 100, 2627 CrossRef CAS PubMed.
- A. Ehlen, C. Wimmer, E. Weber and J. Bargon, Angew. Chem., Int. Ed. Engl., 1993, 32, 110 CrossRef.
- J. Reinbold, K. Buhlmann, K. Camman, A. Wierig, C. Wimmer and E. Weber, Sens. Actuators, B, 1994, 18/19, 77 CrossRef.
- U. Schramm, D. Meinhold, S. Winter, C. Heil, J. Müller-Albrecht, L. Wächter, E. Weber and J. Bargon, Sens. Actuators, B, 2000, 67, 219 CrossRef CAS.
- D. Schindler, F. Eißmann and E. Weber, Org. Biomol. Chem., 2009, 7, 3549 CAS.
- T. Gruber, C. Fischer, W. Seichter, P. Bombicz and E. Weber, CrystEngComm, 2011, 13, 1422 RSC.
-
Bruker, SAINT, SADABS, AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. Sauerbrey, Z. Phys., 1959, 155, 206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The crystallographic information files have been deposited in the Cambridge Structural Database. CCDC 1020736 (1), 1020737 (1a), 1020738 (1b), 1020740 (2a), 1020739 (2b), 1020741 (3A), 1020742 (3B), and 1020743 (3C). The DSC diagrams are included as supplementary figures (Sup. 1 and Sup. 2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce00180c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence