DOI:
10.1039/C4CE02215G
(Paper)
CrystEngComm, 2015,
17, 2754-2768
Architectures varying from discrete molecular units to 2-dimensional coordination polymers and photoluminescence behavior of zinc and cadmium comprising an anionic zwitterion of rigid 4,5-dicarboxy-1,3-dimethyl-1H-imidazolium iodide†
Received
7th November 2014
, Accepted 13th February 2015
First published on 16th February 2015
Abstract
A rigid imidazolium dicarboxylate ligand, 4,5-dicarboxy-1,3-dimethyl-1H-imidazolium iodide (H2DDII), was synthesized and employed in the construction of metal complexes or coordination polymers (CPs) comprising an anionic zwitterion (DDI−), namely, [Zn(Cl)(H2O)2(DDI)]·(H2O) (2), {[Zn2(DDI)3(H2O)2]·I}n (3), [Zn2(DDI)2I]·2H2O (4), [Zn(DDI)2(H2O)3]·(H2O)2 (5) and [Cd(DDI)2]n (6). In addition, the ligand upon crystallization forms a zwitterion, [HDDI, (1)] by losing HI and shows both intra- and intermolecular H-bonding and an interesting intermolecular C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯C short contact. The carboxylate unit of the ligand exhibits terminal monodentate and bidentate chelate modes of coordination. The stoichiometric ratio of metal to ligand, nature of base, type of metal salt and solvent conditions affected the architectures and dimensionality of 2–6 from zero dimensional monomers and dimers to 2D coordination polymers (CPs). Complex 2 forms a discrete molecular unit extended by supramolecular interactions to form 2D rectangular sheets. In 3, a crown shaped macrocycle results in a 2D herringbone CP and extends to a 3D supramolecular architecture on account of a coordination network containing trapped iodide as counter anions. Complex 4 forms a dimer with coordinated iodide ions and forms an infinite 1D ladder shaped supramolecular architecture. Complex 5 forms another discrete molecular unit different from 2, and extends to a 3D architecture through supramolecular interactions. Complex 6 forms a 2D CP with Cd(II) forming an interesting “fish scale pattern” and lacks any counter anion or solvent molecules. Additionally, the thermal stability of complexes 2–6 was analyzed by thermogravimetric analysis. In addition, the solid state photoluminescence properties of H2DDII and compounds 2–6 were investigated at room temperature.
O⋯C short contact. The carboxylate unit of the ligand exhibits terminal monodentate and bidentate chelate modes of coordination. The stoichiometric ratio of metal to ligand, nature of base, type of metal salt and solvent conditions affected the architectures and dimensionality of 2–6 from zero dimensional monomers and dimers to 2D coordination polymers (CPs). Complex 2 forms a discrete molecular unit extended by supramolecular interactions to form 2D rectangular sheets. In 3, a crown shaped macrocycle results in a 2D herringbone CP and extends to a 3D supramolecular architecture on account of a coordination network containing trapped iodide as counter anions. Complex 4 forms a dimer with coordinated iodide ions and forms an infinite 1D ladder shaped supramolecular architecture. Complex 5 forms another discrete molecular unit different from 2, and extends to a 3D architecture through supramolecular interactions. Complex 6 forms a 2D CP with Cd(II) forming an interesting “fish scale pattern” and lacks any counter anion or solvent molecules. Additionally, the thermal stability of complexes 2–6 was analyzed by thermogravimetric analysis. In addition, the solid state photoluminescence properties of H2DDII and compounds 2–6 were investigated at room temperature.
Introduction
The coordination polymers (CPs) or Metal Organic Frameworks (MOFs) constructed by ligands consisting of carboxylate donor groups are one of the main research areas studied over the last two decades.1 CPs comprising carboxylate donor atoms have been employed in a wide range of applications including gas storage, separation, magnetism, luminescence and catalysis.2,3 Multidentate carboxylate donor ligands on account of their diverse coordination modes adopt variable conformations and hence have proven to be excellent candidates for the construction of CPs or MOFs with diverse architectures. This study was extended to the design and synthesis of CPs having N-heterocyclic carboxylate ligands of pyridine, pyrazole and imidazole because of the available mixed multidentate coordination sites including nitrogen and oxygen centers as donor atoms to the metal ions. The presence of an N-heterocyclic unit as well as multiple carboxylate units, with various deprotonated forms, allows for the various coordination modes resulting in different topologies.4–6 In this context, imidazole-4,5-dicarboxylic acid (H3IDC) and its different analogue ligands as linkers have been mainly used for the synthesis of novel CPs.7–10
Functionalized CPs having primary and secondary functional groups have been used as supports and employed in catalysis, wherein the primary functional group coordinates to the metal centre to form an extended network, while the secondary functional group acts as the catalytic site.11 For the past two decades, imidazolium based ligands have been potentially used as precursors for the preparation of N-heterocyclic carbene (NHC) metal complexes in catalysis.12 In addition imidazolium containing systems are known as receptors for anion recognition.13 Recently, a few examples with N-carboxy-functionalized imidazolium salts have been employed in the synthesis of CPs.14 Very recently we have reported the synthesis of backbone thio-functionalized imidazolium salts and their metal complexes as well as the catalytic applications of palladium complexes in catalysis.15
Keeping these facts in mind we have designed a rigid ligand, 4,5-dicarboxy-1,3-dimethyl-1H-imidazolium iodide (H2DDII), containing a backbone functionalized imidazolium salt. The main interest behind the design of this ligand was to use it for the synthesis of metal carboxylate complexes/CPs and further use them as supports to incorporate catalytically active metal centers as well as to study the possible anion trapping inside the cavity. In this regard we have carried out the reaction of H2DDII with (n-Bu3Sn)2O based on the versatile reactivity of organocarboxylic acids with tin(IV) precursors and isolated a hexameric hexagonal organotin 42-membered macrocycle [(n-Bu3Sn)6(μ-L)6(I−)2(MeOH)6] (L = H2DDII) with trapped iodide ions showing an interesting iodide–iodide short contact.16 However if the reactions are carried out with zinc(II) or cadmium(II) precursors, mono or dinuclear zinc complexes or CPs of zinc and cadmium were obtained which were highly influenced by the nature of the metal ion precursors, stoichiometry of the ligand and metal, type of base and solvent conditions. Herein, we report the syntheses and structural characterization of five metal complexes and/or CPS, namely, [Zn(Cl)(H2O)2(DDI)]·H2O (2), {[Zn2(DDI)3(H2O)2]·I}n (3), [Zn2(DDI)2I]·2H2O (4), [Zn(DDI)2(H2O)3]·2H2O (5) and [Cd(DDI)2]n (6) whose structures range from discrete molecular units (monomer and dimer) to 2D CPs and are composed of an anionic zwitterion (DDI−). Besides, we also discuss the structural characterization of the zwitterion [HDDI (1)], obtained upon crystallization of H2DDII, showing intra- as well as intermolecular H-bonding in addition to the intermolecular CO⋯C short contact and resulting in a supramolecular helical 2D network structure. The structural properties of compounds 1–6 have been discussed along with infrared spectra (IR), powder X-ray diffraction (PXRD) and thermogravimetric analysis (TGA). In addition, the solid state fluorescence properties of H2DDII and compounds 2–6 have been investigated.
Experimental section
General details
Anhyd. ZnCl2 and Zn(NO3)2·6H2O were obtained from Alfa Aesar, while Cd(NO3)2·4H2O was obtained from SD Fine, India. 4,5-Imidazoledicarboxylic acid was obtained from Sigma-Aldrich. All other chemicals were used without further purification. Solvents were first dried and then used after distillation. 4,5-Bis(ethoxycarbonyl)-1,3-dimethyl imidazolium iodide was prepared according to the reported literature procedure.17 Microanalyses for all the compounds were recorded using a Perkin Elmer Series-II CHNS/O model 2400 analyzer. IR spectra were recorded on a Bruker model vertex 70 and a Perkin-Elmer model 1320 spectrometer using KBr pellets in the region of 4000–400 cm−1. Thermogravimetric analyses were carried out under N2 atmosphere with a heating rate of 10 °C min−1 using a Mettler Toledo Star System. NMR spectra were recorded on a JEOL ECX-500. Powder X-ray Diffraction (PXRD) data were collected on a PANalytical X'Pert Pro X-ray diffractometer with Cu Kα radiation (λ = 1.540598 Å) at room temperature with a scan step size of 0.02 in 2θ. Solid state emission spectra were recorded using a ZSX primus series spectrometer (Rigaku Corporation) at room temperature. DIAMOND (version 3.0) and Mercury (version 3) were used to view and draw the structures.
4,5-Dicarboxy-1,3-dimethyl-1H-imidazolium iodide (H2DDII)
H2DDII was prepared by the acid hydrolysis of 4,5-bis(ethoxycarbonyl)-1,3-dimethyl imidazolium iodide under reflux conditions for 12 h using 6 N HCl. A clear yellow solution was obtained, which was removed, and the resulting compound was washed with acetone followed by diethyl ether to obtain H2DDII as a light grey compound. Yield: 40% (based on 4,5-imidazoledicarboxylic acid). Anal. calcd. for C7H9N2O4I (312.06 g): C, 26.94; H, 2.91; N, 8.98. Found: C, 26.80; H, 2.90; N, 8.92. IR (KBr, cm−1): 3422 (w), 3145 (m), 3086 (m), 3010 (w), 2979 (w), 2709 (w), 1723 (vs), 1566 (vs), 1473 (vs), 1416 (s), 1370 (m), 1348 (s), 1352 (w), 1300 (s), 1245 (s), 1220 (s), 1144 (vs), 1091 (m), 894 (m), 869 (m), 826 (s), 762 (m), 692 (s), 629 (m), 602 (m), 482 (m). 1H NMR (500 MHz, δ-DMSO-d6): 9.27 (s, 1H, NCHN) and 3.96 (s, 6H, 2 NCH3). 13C-NMR (500 MHz, δ-DMSO-d6) δ 159.1 (2C, CO), 141.1 (NCHN), 129 (2C, CC), 37.7 (2C, NCH3). ESI-MS (+ve) 185 [(M + H)+, 100%] and ESI-MS (−ve) 126.90 (I−).
HDDI (1).
H2DDII was dissolved in DMSO and layered with MeOH. Yellow crystals were obtained after one week. Yield: 63% (based on H2DDII). Anal. calcd. for C7H8N2O4 (184.15 g): C, 45.66; H, 4.38; N, 15.21. Found: C, 45.65; H, 4.30; N, 15.14. IR (KBr, cm−1): 3419 (w), 3139 (w), 3060 (m), 1728 (m), 1573 (vs), 1470 (vs), 1432 (s), 1397 (s), 1371 (s), 1303 (s), 1178 (m), 1024 (m), 907 (w), 869 (w), 833 (w), 770 (m), 657 (br), 629 (m), 600 (w), 498 (w). 1H NMR (500 MHz, DMSO-d6): δ 9.23 (s, 1H, NCHN) and 3.97 (s, 6H, 2 NCH3). 13C-NMR (500 MHz, δ-DMSO-d6) 159.1 (2C, CO), 141.1 (NCHN), 129.1 (2C, CC), 37.6 (2C, NCH3). ESI-MS (+ve) 185 [(M + H)+, 100%].
[Zn(Cl)(H2O)2(DDI)]·H2O (2).
Aq. NH3 (0.1 mL) was added to a solution of H2DDII (46.6 mg, 0.15 mmol) in CH3OH (10 mL) followed by addition of anhyd. ZnCl2 (20.4 mg, 0.15 mmol) and the solution was stirred for 30 min. The solution was allowed to evaporate at room temperature very slowly to afford pale yellow crystals after one day. Yield: 90.2 mg (60% based on anhyd. ZnCl2). Anal. calcd. for C7H13N2O7ClZn (338.03 g): C, 24.87; H, 3.88; N, 8.29. Found: C, 24.83; H, 3.84; N, 8.26. IR (KBr, cm−1): 3425 (s), 3335 (vs), 3128 (m), 3059 (s), 1640 (vs), 1559 (vs), 1474 (s), 1450 (m), 1407 (vs), 1373 (s), 1348 (s), 1321 (vs), 1271 (vs), 1220 (s), 1168 (vs), 899 (s), 880 (w), 826 (s), 802 (m), 723 (m), 698 (m), 632 (s), 617 (s), 474 (s).
Synthesis of {[Zn2(DDI)3(H2O)2]·I}n (3).
Aq. NH3 (0.2 mL) was added to a solution of H2DDII (93.2 mg, 0.3 mmol) in CH3OH (20 mL) followed by addition of ZnCl2 (20.4 mg, 0.15 mmol) as solid. The solution was refluxed for 2 h and allowed to cool to room temperature. Colorless crystals of 3 were obtained after two days by slow evaporation of the solution at room temperature. Yield: 57 mg (91% based on ZnCl2). Anal. calcd. for C21H25N6O14IZn2 (843.15 g): C, 29.91; H, 2.99; N, 9.97. Found: C, 29.90; H, 2.96; N, 9.95. IR (KBr, cm−1): 3325 (m), 3149 (m), 3073 (m), 1644 (vs), 1566 (vs), 1485 (m), 1411 (vs), 1383 (vs), 1348 (vs), 1327 (vs), 1174 (vs), 903 (m), 834 (m), 790 (m), 748 (m), 666 (m), 634 (m), 589 (w), 501 (w), 459 (w).
[Zn2(DDI)2I]·2H2O (4).
Aq. NH3 (0.2 mL) was added to a solution of H2DDII (93.2 mg, 0.3 mmol) in CH3OH (10 mL) followed by addition of Zn(NO3)2·6H2O (37.1 mg, 0.125 mmol). The solution was refluxed for 2 h and allowed to cool to room temperature. Colorless crystals of 4 were obtained after two days by slow evaporation of the solution at room temperature. Yield: 35.1 mg (85% based on Zn(NO3)2·6H2O). Anal. calcd. for C14H18N4O10IZn2 (659.96 g): C, 32.27; H, 3.63; N, 13.72. Found: C, 32.24; H, 3.60; N, 13.69. IR (KBr, cm−1): 3436 (b), 3138 (m), 3064 (s), 2969 (br), 2925 (br), 1733 (m), 1565 (vs), 1470 (vs), 1432 (s), 1414 (m), 1396 (s), 1370 (s), 1300 (vs), 1176 (s), 1090 (s), 910 (w), 869 (m), 838 (m), 770 (s), 680 (w), 629 (s), 605 (w), 497 (m).
Synthesis of [Zn(DDI)2(H2O)3]·2H2O (5).
Triethylamine (0.2 mL) was added to a solution of H2DDII (62.2 mg, 0.2 mmol) in CH3CN/H2O (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 mL) followed by addition of Zn(NO3)2·6H2O (59.4 mg, 0.2 mmol) as solid. The solution was refluxed for 15 h and allowed to cool to room temperature. The solution was allowed to evaporate at room temperature very slowly to afford colorless crystals of 5 after twenty-five days. Yield: 50 mg (25% based on Zn(NO3)2·6H2O). Anal. calcd. for C14H24N4O13Zn (521.76 g): C, 32.23; H, 4.64; N, 10.74. Found: C, 32.22; H, 4.64; N, 10.71. IR (KBr, cm−1): 3414 (m), 3149 (w), 3048 (m), 1630 (vs), 1611 (vs), 1570 (vs), 1474 (s), 1404 (s), 1375 (vs), 1313 (s), 1230 (w), 1175 (s), 1090 (w), 1034 (w), 894 (w), 827 (m), 789 (w), 765 (m), 745 (w), 626 (m), 599 (m), 487 (w), 435 (br), 405 (br).
:8 mL) followed by addition of Zn(NO3)2·6H2O (59.4 mg, 0.2 mmol) as solid. The solution was refluxed for 15 h and allowed to cool to room temperature. The solution was allowed to evaporate at room temperature very slowly to afford colorless crystals of 5 after twenty-five days. Yield: 50 mg (25% based on Zn(NO3)2·6H2O). Anal. calcd. for C14H24N4O13Zn (521.76 g): C, 32.23; H, 4.64; N, 10.74. Found: C, 32.22; H, 4.64; N, 10.71. IR (KBr, cm−1): 3414 (m), 3149 (w), 3048 (m), 1630 (vs), 1611 (vs), 1570 (vs), 1474 (s), 1404 (s), 1375 (vs), 1313 (s), 1230 (w), 1175 (s), 1090 (w), 1034 (w), 894 (w), 827 (m), 789 (w), 765 (m), 745 (w), 626 (m), 599 (m), 487 (w), 435 (br), 405 (br).
Synthesis of [Cd(DDI)2]n (6).
A similar synthetic procedure to that of 5 was performed except that Cd(NO3)2·4H2O (61.6 mg, 0.2 mmol) was used instead of Zn(NO3)2·6H2O. Colorless crystals of 6 were formed after thirty days. Yield: 10 mg (17% based on Cd(NO3)2·4H2O). Anal. calcd. for C14H14N4O8Cd (478.69 g): C, 35.13; H, 2.95; N, 11.70. Found: C, 35.11; H, 2.92; N, 11.65. IR (KBr, cm−1): IR (KBr, cm−1): 3411 (w), 3149 (w), 3085 (m), 1626 (vs), 1607 (vs), 1567 (vs), 1476 (s), 1406 (vs), 1383 (vs), 1357 (s), 1328 (s), 1220 (w), 1175 (s), 1091 (w), 903 (m), 874 (w), 839 (m), 796 (m), 764 (w), 741 (m), 617 (m), 575 (w), 477 (w).
X-ray crystallography
Single crystal X-ray data for 1–6 were collected at 100 K (1–4) and 293 K (5–6) on a Bruker SMART APEX CCD diffractometer using graphite monochromated MoKα radiation (λ = 0.71073 Å). All the structures were solved using SHELXS-97 and refined by full-matrix least squares on F2 using SHELXL-97.18 All hydrogen atoms were included in idealized positions using a riding model. Non-hydrogen atoms were refined with anisotropic displacement parameters. Topological analysis of CPs 3 and 6 was performed using the ADS program of the TOPOS 4.0 Professional structure-topological program package.19
Results and discussion
4,5-Dicarboxy-1,3-dimethyl-1H-imidazolium iodide (H2DDII)
H2DDII was prepared through a stepwise procedure. Firstly, 4,5-bis(ethoxycarbonyl)-1,3-dimethyl imidazolium iodide was synthesized according to a literature procedure through multi-step synthesis (Scheme 1). It was followed by the hydrolysis of 4,5-bis(ethoxycarbonyl)-1,3-dimethyl imidazolium iodide using 6 N HCl, resulting in the formation of ligand H2DDII which was obtained as a grey compound in moderate yield. The formation of H2DDII was confirmed by spectroscopic and spectrometric techniques and it was found to be soluble in DMSO, DMF and MeOH. The complete hydrolysis of 4,5-bis(ethoxycarbonyl)-1,3-dimethyl imidazolium iodide to H2DDII was confirmed through 1H-NMR as indicated by the absence of COOCH2CH3 protons and also by the shielding effect observed for NCHN [9.41 (s, 1H)] and the slight deshielding effect observed for NCH3 [3.90 (s, 6H)] (Fig. S1–S2).
 |
| | Scheme 1 Synthesis of H2DDII. | |
As it was observed in N-carboxy functionalized imidazolium salts, H2DDII is expected to form a zwitterion [HDDI (1)] or an anionic zwitterion (DDI−) (Scheme 2) in solution. HDDI could possibly coordinate to the metal through one or two oxygen atoms of the deprotonated carboxylate moiety, whereas DDI− has four carboxylate O-donor atoms which can possibly coordinate to the metal centers in various fashions such as monodentate terminal/bridging, bidentate chelate/bridging or chelate-bridging20 (Scheme 3), resulting in structures having an anionic zwitterion with different topologies.
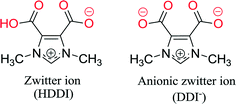 |
| | Scheme 2 Deprotonated forms of H2DDI. | |
 |
| | Scheme 3 Possible coordination modes of the anionic zwitterion (DDI−). | |
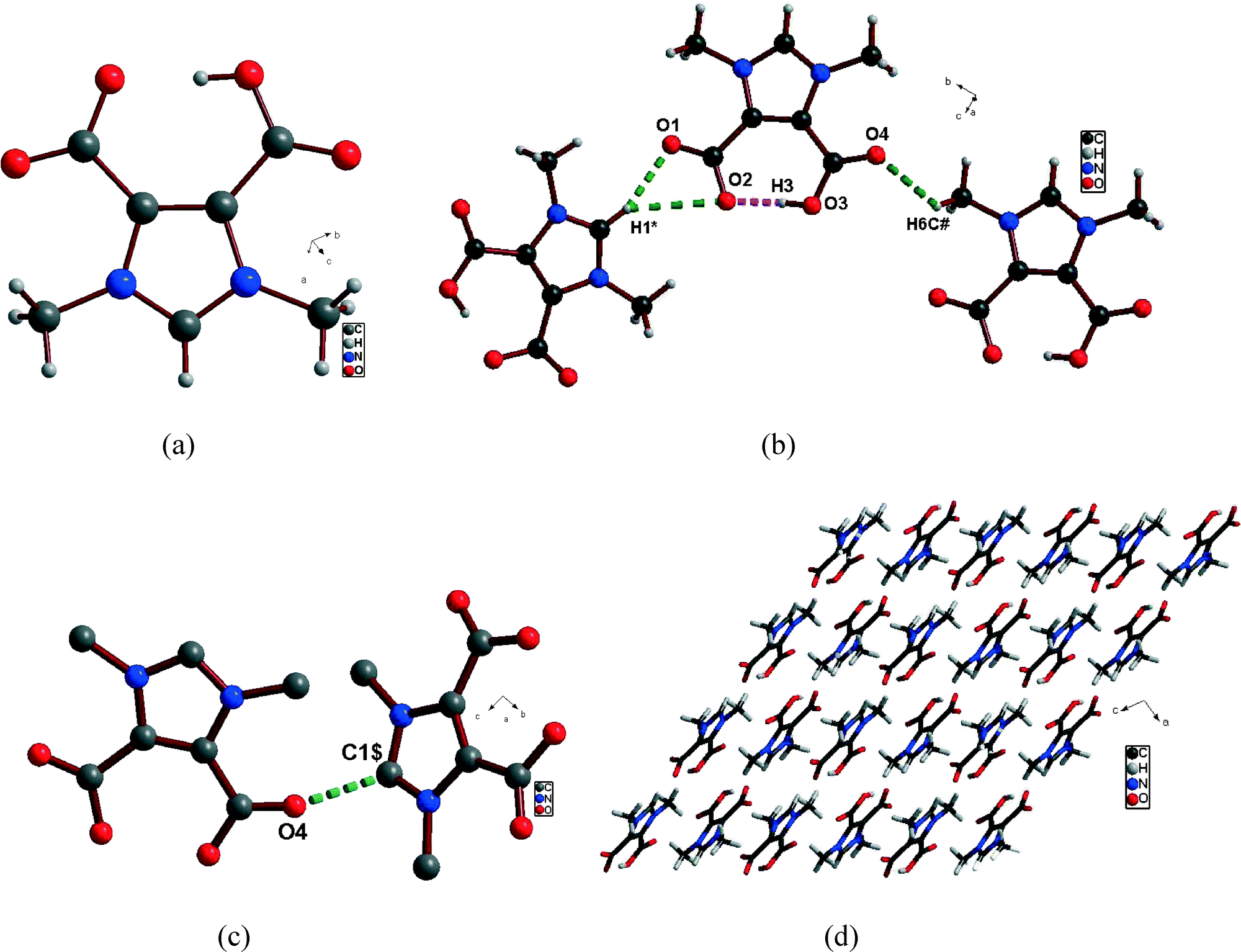
Interestingly, H2DDII upon crystallization in DMSO and methanol formed HDDI (1), which was characterized by IR, NMR, ESI-MS and single crystal X-ray diffraction methods (Fig. S3–S4). 1 crystallizes in the monoclinic system with the space group P21/n (Table 1) and was found to have a zwitterionic structure with the loss of HI (Fig. 1a) in solution. In 1, an intramolecular H-bond exists between O2⋯H3–O3, arising from the carboxylate units, with a distance of 1.587(2) Å (Fig. 1b).21 In addition, intermolecular O⋯H–C interactions22 exist between O1⋯H1*–C1 as well as O2⋯H1–C1 with adjacent HDDI units, while O4⋯H6C#–C6 interactions were present between the stacked up HDDI units, one over the other layers (Fig. 1b, Table 3). Besides, there also exists an interesting intermolecular CO⋯C short contact between C5O4⋯C1$ having a distance of 2.895(3) Å and connecting two adjacent HDDI units (Fig. 1c, Table 3).23 These interactions collectively tend to form perpendicular adjacent layers of HDDI and also stack up one HDDI unit over the other, leading to an extended supramolecular helical 2D network structure (Fig. 1d).
Table 1 Crystallographic data and structural refinements for 1–6
|
|
1
|
2
|
3
|
4
|
5
|
6
|
| Formula |
C7H8N2O4 |
C7H13N2O7ClZn |
C21H25IN6O14Zn2 |
C14H18IN4O10Zn2 |
C14H24N4O13Zn |
C14H14CdN4O8 |
| Fw |
184.15 |
338.01 |
843.11 |
659.96 |
521.74 |
478.69 |
| Crystal system |
Monoclinic |
Monoclinic |
Orthorhombic |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/n |
Pccn
|
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
C2/c |
|
a (Å) |
7.052(2) |
8.4910(17) |
22.552(5) |
7.9324(13) |
15.075(2) |
8.8373(8) |
|
b (Å) |
8.324(3) |
8.0410(16) |
10.123(2) |
8.4897(14) |
8.3079(12) |
13.3995(8) |
|
c (Å) |
13.410(4) |
18.847(4) |
13.594(3) |
10.8444(18) |
16.676(3) |
13.4110(10) |
|
α (deg) |
90 |
90 |
90 |
80.027(3) |
90 |
90 |
|
β (deg) |
102.299(6) |
101.16(3) |
90 |
72.510(3) |
92.800(3) |
91.947(2) |
|
γ (deg) |
90 |
90 |
90 |
64.679(2) |
90 |
90 |
|
V (Å3) |
769.1(4) |
1262.5(4) |
3103.4(1) |
628.8(2) |
2086.1(5) |
1587.2(2) |
|
Z
|
4 |
4 |
4 |
1 |
4 |
4 |
|
ρ
calc (g cm−3) |
1.590 |
1.778 |
1.804 |
1.743 |
1.661 |
2.003 |
|
μ/mm−1 |
0.132 |
2.184 |
2.614 |
3.185 |
1.253 |
1.434 |
|
F(000) |
384 |
688 |
1672 |
323 |
1080 |
952 |
|
T (K) |
100(2) |
100(2) |
100(2) |
100(2) |
293(2) |
293(2) |
|
θ
min/max range (°) |
2.90–26.00 |
2.20–25.99 |
1.81–26.00 |
2.66–26 |
1.35–26.00 |
2.76–26.00 |
| Reflections collected/unique |
4130/1504 |
6839/2461 |
16258/3049 |
3533/2413 |
11326/4082 |
4288/1532 |
| Data/restraints/parameter |
1504/0/121 |
2461/9/183 |
3049/3/208 |
2413/3/164 |
4082/15/324 |
1532/6/125 |
| GOF on F2 |
1.165 |
1.071 |
1.035 |
1.091 |
1.143 |
1.252 |
|
R
int
|
0.0368 |
0.0460 |
0.0590 |
0.0189 |
0.0464 |
0.0311 |
|
R
1; wR2[I > 2σ(I)] |
0.0486; 0.1357 |
0.0397;0.1079 |
0.0527; 0.1418 |
0.0534; 0.1627 |
0.0516; 0.1324 |
0.0316; 0.0752 |
|
R
1; wR2 (all data) |
0.0602; 0.1669 |
0.0455; 0.1135 |
0.0683; 0.1576 |
0.0595; 0.1792 |
0.0713; 0.1726 |
0.0366; 0.1078 |
 |
| | Fig. 1 (a) The zwitterionic structure of 1. (b) Inter-(green color) and intramolecular (pink color) H-bonding interactions in 1. (c) Intermolecular CO⋯C short contact in 1. (d) Extended supramolecular architecture of 1. [H1* is at −1/2 + x, 1/2 − y, −1/2 + z; H6C# is at x, 1 + y, z; C1$ is at 1/2 − x, 1/2 + y, 1/2 + z]. | |
Syntheses of 2–6
The coordination ability of H2DDII with the metal ions was evaluated by the synthesis of complexes of Zn(II) and Cd(II) using a simple evaporation technique. The removal of the carboxylic acid proton from H2DDII was facilitated by the use of aq. NH3 and Et3N as base. In these reactions, the pH of the solution was maintained always between 8 and 9. The syntheses of complexes 2–4 were accomplished by using ZnCl2 (2 and 3) and Zn(NO3)2·6H2O (4) as metal precursors and aq. NH3 as base in methanol (Scheme 4). The stoichiometry of the metal and ligand plays an important role in determining the structure of the resulting products. The stoichiometric ratio of ZnCl2 to H2DDII of 1:1 and 1:2 yields 2 and 3, respectively. The same synthetic conditions as those for 2 and 3 were followed for the preparation of 4, except that Zn(NO3)2·6H2O was used in the reaction, whereas the syntheses of 5 and 6 were carried out using a strong base and M(NO3)2·xH2O (M = Zn (5), x = 6; M = Cd (6), x = 4) in CH3CN/H2O medium (Scheme 4). All the products (2–6) were insoluble in common organic solvents. The IR spectra of 2–6 showed stretching frequencies for νas (COO) and νs (COO) around 1600 cm−1 and 1400–1300 cm−1, respectively, which support the binding of carboxylate to the metal ion. Further, the absence of strong IR bands around 1700 cm−1 in 2–6 and the presence of multiple stretching frequency peaks in the region of metal carboxylates suggest variable binding modes of carboxylate units to metal ions. To understand better the product formation and the details of the final structures of products 2–6, a single crystal X-ray diffraction study was carried out.
 |
| | Scheme 4 Synthetic scheme and structures of 1–6. | |
Solid state structures of 2–6
[Zn(Cl)(H2O)2(DDI)]·H2O (2).
Single crystal X-ray diffraction studies shows that 2 crystallizes in the monoclinic space group P21/n (Table 1). It consists of a Zn(II) ion, one DDI− unit, one Cl− ion and two coordinated H2O molecules along with one H2O molecule as water of crystallization. The Zn(II) ion lies in a tetrahedral environment and is coordinated by one donor oxygen atom provided by the DDI− unit and one chloride ion, while the remaining two sites are occupied by two H2O molecules (Fig. 2a). In 2, both the hydrogen atoms from carboxylates along with the iodide ion are eliminated. The O-atom from the carboxylate unit of DDI− coordinates to Zn(II) in monodentate terminal mode (I; Scheme 3), while the second carboxylate unit has a free oxygen-donor site. Hence, the charge is balanced since one of the carboxylate groups remains uncoordinated. Thus, 2 can be described as an anionic zwitterion zinc complex as it was also observed in N-carboxy imidazolium metal dicarboxylates.14
 |
| | Fig. 2 (a) The coordination environment around Zn(II) ions in 2. (b) H-bonding interactions in 2. [H7C* is at −1/2 + x, 3/2 − y, 1/2 + z; H66# is at 3/2 − x, −1/2 + y, 1/2 − z]. (c) Single unit of a rectangular sheet in 2. (d) 2D rectangular sheet in 2. | |
The Zn–O bond distance of carboxylate oxygen to zinc in 2 is found to be 2.009(4) Å (Zn–O1), comparable to the bond lengths reported with such a ligand system,9h whereas those for the water molecules coordinated to the Zn(II) ion are nearly the same [av. 2.001(3) Å], while the bond distance of Zn–Cl is 2.274(11) Å (Table 2). The asymmetric unit of 2 extends further through the H-bonding existing between the chloride ion and H7C* (from N–CH3) and H66# (from H2O) (Fig. 2b, Table 3) to form a 2D rectangular sheet (Fig. 2c and d). Each rectangular sheet comprises four monomeric units of 2 in which the two DDI− units are involved in facilitating the extended structure formation. The orientation of DDI− (along a-axis) within the rectangular sheet and in the 2D layers is anti-parallel to each other. The chloride ions are oriented in opposite directions between two layers, but are in the same direction to the ones adjacent to it. The orientations of DDI− and Cl− ions play a significant role in extending the monomer through H-bonding. The distances between Zn(II) ions in such a rectangular sheet were found to be 4.410(9) Å and 11.481(3) Å for the stacked up and adjacent one, respectively (Fig. S5). The 2D rectangular sheets are further connected via the lattice water molecules which tend to act as a bridge through CH⋯O interactions, (Table 3, Fig. S6), leading to the formation of a 3D supramolecular architecture (Fig. S7).
Table 2 Selected bond distances (Å) and angles (deg) for compounds 2–6a
| [Zn(Cl)(H2O)2(DDI)]·H2O (2) |
{[Zn2(DDI)3(H2O)2]·I}n (3) |
[Zn2(DDI)2I]·2H2O (4) |
[Zn(DDI)2(H2O)3]·2H2O (5) |
[Cd(DDI)2]n (6) |
|
Symmetry codes for 3 #1 −x + 1/2, −y + 1/2, z #2 −x, y + 1/2, −z + 1/2 #3 −x, y − 1/2, −z + 1/2; 4 #1 −x + 1, −y + 2, −z + 1; 6 #1 x − 1/2, y − 1/2, z #2 x + 1/2, y + 1/2, z #3 −x + 2, y, −z + 1/2.
|
| Zn(1)–Cl1 2.2743(11) |
Zn(1)#2–O2 2.034(4) |
Zn(1)–I(1) 2.2733(14) |
Zn(1)–O(1) 2.073(3) |
Cd(1)–O(1) 2.248(3) |
| Zn(1)–O(1) 2.009(2) |
Zn(1)–O3 1.985(3) |
Zn(1)–I(2) 2.2564(13) |
Zn(1)–O(5) 2.007(3) |
Cd(1)–O(3) 2.417(3) |
| Zn(1)–O(1W) 2.002(3) |
Zn(1)–(O1W) 2.041(5) |
Zn(1)–O(1) 1.992(4) |
Zn(1)–O(1W) 2.086(3) |
Cd(1)–O(4) 2.438(3) |
| Zn(1)–O(2W) 1.999(3) |
Zn(1)–O7 1.999(3) |
Zn(1)#1–O(4) 1.966(4) |
Zn(1)–O(2W) 2.023(3) |
O(1)–Cd(1)–O(3) 94.65(12) |
| O(2W)–Zn(1)–O(1W) 117.29(11) |
O(3)–Zn(1)–O(7) 104.95(14) |
Zn(1)–O(4)#1 1.967(4) |
Zn(1)–O(3W) 2.055(3) |
O(1)–Cd(1)–O(4) 82.13(12) |
| O(2W)–Zn(1)–O(1) 105.94(10) |
O(3)–Zn(1)–O(2)#3 107.77(15) |
O(4)#1–Zn(1)–O(1) 101.94(15) |
O(5)–Zn(1)–O(2W) 108.26(14) |
O(3)–Cd(1)–O(4) 54.28(11) |
| O(1W)–Zn(1)–O(1) 105.59(10) |
O(7)–Zn(1)–O(2)#3 95.76 |
O(4)#1–Zn(1)–I(2) 105.84(11) |
O(5)–Zn(1)–O(3W) 92.23(13) |
O(1)–Cd(1)–O(1)#3 117.04(18) |
| O(2W)–Zn(1)–Cl(1) 113.46(8) |
O(3)–Zn(1)–O(1W) 123.10(17) |
O(1)–Zn(1)–I(2) 112.69(11) |
O(2W)–Zn(1)–O(3W) 97.06(13) |
O(1)#3–Cd(1)–O(3) 109.58(13) |
| O(1W)–Zn(1)–Cl(1) 109.15(8) |
O(7)–Zn(1)–O(1W) 113.66(18) |
O(4)#1–Zn(1)–I(1) 122.35(11) |
O(5)–Zn(1)–O(1) 150.98(14) |
O(3)–Cd(1)–O(3)#3 133.04(16) |
| O(1)–Zn(1)–Cl(1) 104.20(7) |
O(2)#3–Zn(1)–O(1W) 108.08(17) |
O(1)–Zn(1)–I(1) 103.03(12) |
O(2W)–Zn(1)–O(1) 100.08(13) |
O(1)#3–Cd(1)–O(4) 157.57(13) |
|
|
|
I(2)–Zn(1)–I(1) 110.74(5) |
O(3W)–Zn(1)–O(1) 90.36(13) |
O(3)#3–Cd(1)–O(4) 89.03(11) |
|
|
|
|
O(5)–Zn(1)–O(1W) 86.43(13) |
|
|
|
|
|
O(2W)–Zn(1)–O(1W) 92.09(13) |
|
|
|
|
|
O(3W)–Zn(1)–O(1W) 170.70(13) |
|
|
|
|
|
O(1)–Zn(1)–O(1W) 86.41(12) |
|
Table 3 Hydrogen bonding interaction parameters in 1–6
| (D–H⋯A) |
d(H⋯A) (Å) |
d(D⋯A) (Å) |
<(A⋯H–D)(°) |
|
1
|
| C1–H1*⋯O1 |
2.22 |
3.13(4) |
161 |
| C1–H1*⋯O2 |
2.69 |
3.52(1) |
146 |
| C6–H6C#⋯O4 |
2.57 |
3.33(2) |
134 |
| O3–H3⋯O2 |
1.58 |
2.42(6) |
177 |
| C5O4⋯C1$ |
2.89(5) |
|
133 |
|
2
|
| O2W–H66#⋯Cl1 |
2.66 |
3.46(5) |
151 |
| C7–H7C*⋯Cl1 |
2.69 |
3.61(9) |
163 |
| O3W–H78⋯O4 |
2.10 |
2.92(1) |
171 |
| C1–H1⋯O3W |
2.28 |
3.12(7) |
150 |
| C6–H6A⋯O3W |
2.58 |
3.44(5) |
150 |
|
3
|
| O5–H15⋯I1# |
3.01 |
3.84(1) |
162 |
| C9–H9B⋯I1# |
3.12 |
4.00(4) |
153 |
|
1. H1* is at −1/2 + x, 1/2 − y, −1/2 + z; H6C# is at x, 1 + y, z; C1$ is at 1/2 − x, 1/2 + y, 1/2 + z. 2. H7C* is at −1/2 + x, 3/2 − y, 1/2 + z; H66# is at 3/2 − x, −1/2 + y, 1/2 − z]. 3. I1# is at 1/2 − x, −1 + y, −1/2 + z. |
|
4
|
| C1–H1⋯I2 |
2.55 |
3.40(3) |
150 |
| C6–H6C⋯I2 |
2.79 |
3.68(9) |
152 |
|
5
|
| O4W–H120⋯O6 |
1.88 |
2.67(7) |
166 |
| O5W–H131⋯O7 |
2.06 |
2.88(1) |
176 |
| C14–H14A⋯O7 |
2.69 |
3.56(2) |
151 |
| O1W–H9⋯O7 |
1.98 |
2.75(1) |
158 |
| O2W–H100⋯O8 |
1.96 |
2.70(9) |
152 |
|
6
|
| C1–H4⋯O2 |
2.20 |
2.97(4) |
140 |
| C7–H5C⋯O3 |
2.41 |
3.28(9) |
152 |
{[Zn2(DDI)3(H2O)2]·I}n (3).
Single crystal X-ray diffraction studies show that 3 crystallizes in the orthorhombic space group Pccn (Table 1). The analysis of the crystal structure reveals that 3 is an anionic zwitterion 2D CP with a basic unit composed of two zinc, three DDI− units, two water molecules and an iodide as a counter anion, with a 2-fold imposed symmetry where the C10–H10 bond and I1 lie on different 2-fold axes. Each Zn(II) ion lies in a tetrahedral environment and is coordinated by three donor oxygen-atoms provided by three individual DDI− units and one H2O molecule occupies the fourth coordination site (Fig. 3a). All the three oxygen atoms of DDI− coordinate to the Zn(II) ion in monodentate terminal mode (II; Scheme 3). The Zn–O bond distance in 3 of carboxylate oxygen atoms to zinc falls in the range of 1.985(3) Å to 2.041(5) Å and this is consistent with the already reported values in the literature,9h while the bond angles around zinc vary from 95.76(14)° to 123.10(17)°, hence the geometry of zinc can be described as a distorted tetrahedron (Table 2). In 3, the oxygen atoms from the second carboxylate unit of DDI− bind to the other zinc atoms to form a hexameric 42-membered crown-shaped macrocycle ring (Fig. 3b). The macrocycle consists of six Zn(II) atoms which are connected through six DDI− in the same direction while the other two (breadthwise) are in the opposite direction and hence form the crown shape of the macrocycle. Further, the O-atoms of the other carboxylate group of DDI− units from the hexamer coordinate to another Zn(II) ion, which extends to form a 2D anionic zwitterion zinc CP with a herringbone network (Fig. 3c). Through topological analysis, 3 can be represented as a 63 hcb topology with the Zn centre as a 3-connected uninodal net (Fig. 3d). The iodide ions in 3 are trapped inside the quasi hydrophilic channels of the CP and are involved in the H-bonding between I1#⋯H9B (from N–CH3) and I1#⋯H15 (from H2O) (Fig. 3e, Table 3). The 2D CP is further connected through an interlayer CH⋯O interaction between CH3 and CH of imidazolium units resulting in the formation of a 3D supramolecular network (Fig. 3f).
 |
| | Fig. 3 (a) The coordination environment around Zn(II) ions in 3 (hydrogen atoms are omitted for clarity). (b) Hexameric 42-membered crown-shaped macrocycle ring in 3. (c) 2D herringbone pattern in 3. (d) Topological representation of 3-connected nodes in 3. (e) H-bonding interactions in 3 [I1# is at 1/2 − x, −1 + y, −1/2 + z]. (f) 3D supramolecular network of 3 with iodide ions. | |
[Zn2(DDI)2I]·2H2O (4).
Single crystal X-ray analysis shows that 4 crystallizes in the triclinic space group P (Table 1), and forms an anionic zwitterion zinc dimer consisting of two Zn(II) ions, two DDI− units, four iodide ions (one-fourth occupancy) and two lattice water molecules. Both Zn(II) ions lie in a distorted tetrahedral environment (bond angles vary from 101.94° to 122.35°), connected by two individual DDI− units. Each DDI− unit provides two donor oxygen atoms to form a 14-membered macrocyclic ring, while the remaining four sites are occupied by the four iodide ions (Fig. 4a). Unlike 3, in complex 4, the iodide ions are coordinated to the zinc ion terminally and form a dimer, hence thwarting the formation of an extended CP. The oxygen atoms of the DDI− unit are coordinated to zinc in a monodentate terminal fashion (II; Scheme 3), with bond distances of 1.992(4) Å and 1.966(4) Å for Zn–O1 and Zn–O4* respectively,9h whereas the bond distances of Zn–I are 2.273(14) Å and 2.256(13) Å for Zn–I1 and Zn–I2 respectively.24 Within the macrocycle, the orientations of the two DDI− units are anti-parallel to each other, while the iodide ions are perpendicular to the macrocycle. The orientation of the DDI− units and iodide ions facilitates the H-bonding interactions between two neighboring dimer units [I2⋯H1–C1 (imidazolium) and I2⋯H6C (N-methyl) (Fig. S9; Table 3)]. These supramolecular interactions between adjacent dimer units lead to an infinite ladder-shaped 1D supramolecular network (Fig. 4b). The Zn(II) ions form the connections of the ladder and the distance between Zn⋯Zn in a ladder is 4.840(1) Å along the rung and 8.489(2) Å along the rail (Fig. S10).
 |
| | Fig. 4 (a) The coordination environment around Zn(II) ions in 4. [O3* and O4* are at equivalent positions 1 − x, 2 − y, 1 − z]. (b) Infinite ladder-shaped 1D supramolecular network of 4. | |
[Zn(DDI)2(H2O)3]·2H2O (5).
It was observed that using aq. NH3 doesn't always facilitate the complete removal of HX (Cl− or I−) and hence the expected CPs probably were not observed. Keeping this in mind Et3N which is a stronger base as compared to aqueous NH3 was used in the reaction medium. Thus, complex 5 was obtained by using a stoichiometric ratio of 1:1 of the ligand and Zn(NO3)2·6H2O, using Et3N as base in a CH3CN–H2O mixture at room temperature.
The same reaction procedure using ZnCl2 was not successful. Single crystal X-ray diffraction shows that 5 crystallizes in the monoclinic space group P21/c (Table 1), and consists of one Zn(II) ion, two DDI− units, three coordinated H2O molecules and two lattice H2O molecules. Zn(II) lies in an octahedral environment and three of the coordination sites are occupied by three O-atoms from the two DDI− units. Interestingly, in this case two coordination modes of DDI− were observed viz. monodentate terminal and bidentate chelate. One of the DDI− units coordinates to Zn(II) in a monodentate terminal fashion, while the second unit of DDI− shows bidentate chelate mode (III; Scheme 3). The remaining three sites are occupied by three H2O molecules (Fig. 5a). Like 1 and 2, a loss of HI is observed in 5, however the final product obtained here is once again an anionic zwitterion zinc complex formed by DDI−.
 |
| | Fig. 5 (a) The coordination environment around Zn(II) ions in 5. (b) 18-membered macrocycle of 5. (c) 2D supramolecular network of 5 (H-bonds are omitted for clarity). | |
The Zn–O bond length in 5 is found to be in the range of 2.007(3) Å to 2.086 (3) Å which is comparable to literature values.9h The bond angles in 5 vary from 86.41(12)° to 170.70(13)° (Table 2). Two of the monomer units are connected to each other by H-bonding and form an 18-membered macrocycle (Fig. 5b). Each of these macrocycles consists of two DDI− units coordinated in monodentate terminal mode from two adjacent monomers, while the remaining two DDI− units coordinated in bidentate chelate mode lack any such supramolecular interactions. These monomer units along with the coordinated and lattice H2O molecules further extend the macrocycle through CH⋯O interactions and H-bonds (Table 3, Fig. 5c) and result in a 2D supramolecular network.
[Cd(DDI)2]n (6).
In order to evaluate the metal ion effect, the reaction was carried out using cadmium nitrate and NEt3 under similar conditions to those of 5. The same synthetic procedure as that for 2–4 was not fruitful in this case and performing the same reaction by using CdCl2 was not successful. Single crystal X-ray diffraction shows that 6 crystallizes in the monoclinic space group C2/c (Table 1). In 6, each Cd(II) ion lies in a twofold axis and is in a distorted octahedron environment coordinated by six donor O-atoms provided by four anionic DDI− units (Fig. 6a). Similar to 5, in 6 two coordination modes of DDI−, i.e. monodentate terminal and bidentate chelate, were observed. Two of the DDI− units are coordinated to the Cd(II) ion in bidentate chelate mode, while the remaining two DDI− units are coordinated in monodentate terminal mode (III; Scheme 3). The Cd–O bond distance in 6 falls in the range of 2.417(3) Å to 2.248(3) Å, all of which are comparable to those reported for other imidazole-based dicarboxylate Cd(II) complexes (Table 2).9h,i The DDI− units, which are coordinated to one Cd(II) ion in bidentate chelate mode, are further coordinated to another Cd(II) ion in monodentate terminal mode and vice versa. Thus, there exists a tetrameric 28-membered macrocyclic ring (Fig. 6b). Each macrocycle consists of four Cd(II) ions which are coordinated to four DDI− units and the distance between two Cd(II) centers was found to be 8.025(6) Å. The four DDI− units are oriented perpendicular to each other. Further, each of these tetramer units in 6 extends through the remaining carboxylate oxygen atoms forming another tetramer and results in a 2D anionic zwitterion cadmium CP showing an interesting “fish scale pattern” (Fig. 6c). Through topological analysis, 6 can be represented as a 4462 sql topology with the Cd centre as a 4-connected uninodal net (Fig. 6d). The 2D CP is further connected through the intermolecular CH⋯O interactions existing between carboxylate oxygen with different H-donor carbon atoms, to form a 3D supramolecular network (Fig. 6e). The 3D supramolecular network shows packing in an ABAB fashion with the adjacent layer (Table 3, Fig. S11 and S12).
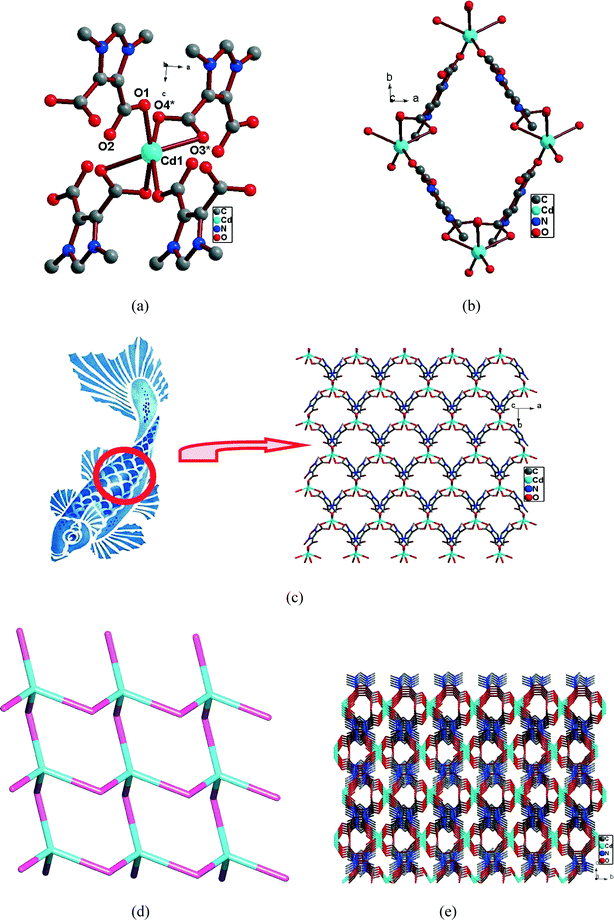 |
| | Fig. 6 (a) The coordination environment around Cd(II) ions in 6 (hydrogen atoms are omitted for clarity). [O3* and O4* are at equivalent positions 3/2 − x, 1/2 + y, 1/2 − z]. (b) Tetrameric 28-membered macrocyclic ring of 6. (c) Fish scale pattern of 2D CP 6. (d) Topological representation of 4-connected nodes in 6. (e) 3D supramolecular structure of 6. | |
Thermogravimetric and powder X-ray diffraction analyses
The thermal stability of 2–6 was studied using thermogravimetric analysis (TGA) (Fig. 7). The TGA curve of 2 shows an initial weight loss of 26.2% (calculated 26.46%) up to 211 °C, corresponding to the loss of one chloride ion, two coordinated water molecules and one lattice water molecule. At 255 °C, 2 shows a gradual decrease in weight due to continuous loss of the ligand (residue 28.4%). The TGA curve of 3 shows an initial weight loss of 3.3% (calculated 2.13%) at 96.68 °C corresponding to the loss of one coordinated water molecule. The second weight loss of 27% (calculated 22%) at 149 °C corresponds to loss of one of the coordinated ligands. The third weight loss of 42% (calculated 43.9%) at 262 °C shows the loss of the remaining two coordinated ligands (residue 27.7%). The TGA curve of 4 shows that it is stable up to 180 °C and shows an initial weight loss of 32.7% (calculated 33.49%) at 185 °C, corresponding to the loss of one ligand and two lattice water molecules. At 250 °C, 4 shows a gradual weight loss due to the loss of one ligand unit and iodide ions (residue 20.7%). The TGA curve of 5 shows that it is stable up to 193 °C and shows a sudden weight loss of 90% at 250 °C, corresponding to the loss of the coordinated ligands and water molecules (residue 10%). The TGA curve of 6 shows that it is stable up to 200 °C and shows a gradual weight loss of 62.91% (calculated 64.68%) corresponding to the loss of four ligand units which are coordinated to Cd(II) (residue 37.09%).
 |
| | Fig. 7 TGA curves of compounds 2–6. | |
To confirm the sample purity, powder X-ray diffraction was performed on compounds 1–6 and it was observed that the simulated spectra correlate with the experimental spectra, which confirms the phase purity of these compounds at room temperature (Fig. S13). To confirm the stability of complexes 2–6 at high temperature, variable temperature PXRD was also performed and it was observed that all the complexes are stable up to 100 °C. Complex 3 was found to be stable even after heating at 160 °C, while complexes 4–6 were found to be stable even at 200 °C. To check the reversibility of water removal in CP 3, it was first heated at 100 °C and then re-immersed in water; it was found that the PXRD pattern of the re-immersed compound does not match with that of CP 3 confirming the irreversible nature of water removal (Fig. S14).
Photoluminescence
CPs consisting of transition metals with a d10 electronic configuration such as Zn and Cd and suitable organic chromophores are found to exhibit excellent photoluminescence properties.25 Therefore the complexes synthesized by using these metal ions can be employed as new luminescent materials. Consequently, the photoluminescence properties of complexes 2–6 were investigated at room temperature (Fig. 8). In the solid state, unlike imidazole-4,5-dicarboxylic acid (H3IDC) and its different analogue ligands which show moderate to weak luminescence, the free H2DDII shows intense luminescence with emission maximum at 348 nm (λex 315 nm) due to the π*–π transition as well as XLCT (halide to ligand charge transfer) due to iodide ions.8 Compounds 2–4 are red shifted from the ligand and exhibit intense luminescence emission at 397 nm (λex 250 nm), 396 nm (λex 250 nm) and 395 nm (λex 265 nm), respectively. A similar red shift in the luminescence emission as well as fluorescence quenching, due to a decrease in intensity, was observed in compounds 5 and 6 at 398 nm (λex 255 nm) and 422 nm (λex 365 nm), respectively, as compared to the ligand. The cause of red shift for compounds 2–6 can be attributed to ligand to metal charge transfer (LMCT) transition.8c,25c The difference in the emissive properties of complexes 2–4 as compared to 5–6 having the same ligand could be due to the presence of anions, Cl− (2) and I− (3 and 4), and the difference in the coordination environment around the metal ion and variable architectures.25c,26 The anion plays an important role in the fluorescence properties of these complexes by enhancing the intensity by XLCT transition, which consequently reduces the probability of fluorescence quenching. On the removal of the anion in 5, in spite of having the same metal ion, Zn(II), and ligand as those of 2–4, a large decrease in the fluorescence intensity was observed due to the absence of XLCT transition. The quenching in 6 can be explained by the absence of XLCT and by taking into consideration the presence of the heavy atom perturbation effect due to Cd which decreases the luminescence intensity due to non-radiative energy dissipation.27
 |
| | Fig. 8 Solid state photoluminescence spectra of compounds 2–6. | |
Conclusions
In summary, we have designed a rigid imidazolium dicarboxylate ligand, H2DDII, which upon crystallization forms the zwitterionic structure HDDI (1). Five anionic zwitterion metal complexes and/or CPs were synthesized, namely, [Zn(Cl)(H2O)2(DDI)]·H2O (2), {[Zn2(DDI)3(H2O)2]·I}n (3), [Zn2(DDI)2I]·2H2O (4), [Zn(DDI)2(H2O)3]·2H2O (5) and [Cd(DDI)2]n (6), exhibiting different architectures ranging from discrete molecular units to 2D CPs. Complexes 2 and 5 were monomeric units, while 4 was a dimer which formed an infinite 1D ladder through supramolecular interactions. CP 3 and CP 6 resulted in 2D CPs with herringbone and fish scale patterns, respectively. The stoichiometric ratio of metal to ligand, nature of metal salt, solvent conditions and nature of base influenced the structure of the resulting complexes. The iodide ion is present either as a coordinating anion to the metal centre or captured inside the CP as a counter anion. Further, the solid state photoluminescence properties of H2DDII and compounds 2–6 were also investigated. Compared to the imidazole or substituted imidazole dicarboxylic acids strong emission was exhibited by H2DDII and compounds 2–6. Employing these complexes/CPs as support systems is in progress.
Acknowledgements
The authors thank the Council of Scientific and Industrial Research (CSIR), the Department of Science and Technology (DST), the Government of India for financial support and the Indian Institute of Technology (IIT), Kanpur, for infrastructural facilities. We also thank “Thematic Unit of Excellence on Soft Nanofabrication with Application in Energy, Environment and Bioplatform at IIT Kanpur” and PG Research lab, Department of Chemical Engineering, IIT Kanpur, for the PXRD facility. ST thanks CSIR for the research fellowship. We are thankful to Mr. Musheer Ahmad for helping us by drawing the nets using TOPOS.
References
-
(a) Y.-B. Zhang and J.-P. Zhang, Pure Appl. Chem., 2013, 85, 405 CAS;
(b) R. L. LaDuca, Coord. Chem. Rev., 2009, 253, 1759 CrossRef CAS PubMed;
(c) D. J. Tranchemontagne, J. L. Mendoza-Cortés, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC;
(d) S. I. Vagin, A. K. Ott and B. Rieger, Chem. Ing. Tech., 2007, 79, 767 CrossRef CAS;
(e) N. Guillou, C. Livage and G. Férey, Eur. J. Inorg. Chem., 2006, 4963 CrossRef CAS;
(f) W. Mori, S. Takamizawa, C. N. Kato, T. Ohmura and T. Sato, Microporous Mesoporous Mater., 2004, 73, 31 CrossRef CAS PubMed;
(g) A. Erxleben, Coord. Chem. Rev., 2003, 246, 203 CrossRef CAS;
(h) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed;
(i) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS PubMed.
-
(a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed;
(b) Z.-J. Lin, L.-W. Han, D.-S. Wu, Y.-B. Huang and R. Cao, Cryst. Growth Des., 2013, 13, 255 CrossRef CAS;
(c) J. Sahu, M. Ahmad and P. K. Bharadwaj, Cryst. Growth Des., 2013, 13, 2618 CrossRef CAS;
(d) Y.-S. Xue, F.-Y. Jin, L. Zhou, M.-P. Liu, Y. Xu, H.-B. Du, M. Fang and X.-Z. You, Cryst. Growth Des., 2012, 12, 6158 CrossRef CAS;
(e) Y.-L. Gai, K.-C. Xiong, L. Chen, Y. Bu, X.-J. Li, F.-L. Jiang and M.-C. Hong, Inorg. Chem., 2012, 51, 13 CrossRef PubMed;
(f) K. S. Jeong, Y. B. Go, S. M. Shin, S. J. Lee, J. Kim, O. M. Yaghi and N. Jeong, Chem. Sci., 2011, 2, 877 RSC;
(g) M. Du, C.-P. Li, J.-M. Wu, J.-H. Guo and G.-C. Wang, Chem. Commun., 2011, 47, 8088 RSC.
-
(a) J.-Y. Wua, C.-W. Yang, H.-F. Chen, Y.-C. Jao, S.-M. Huang, C. Tsai, T.-W. Tseng, G.-H. Lee, S.-M. Peng and K.-L. Lu, J. Solid State Chem., 2011, 184, 1740 CrossRef PubMed;
(b) M. A. Nadeem, M. Bhadbhade, R. Bircher and J. A. Stride, Cryst. Growth Des., 2010, 10, 4060 CrossRef CAS;
(c) G.-Q. Kong and C.-D. Wu, Cryst. Growth Des., 2010, 10, 4590 CrossRef CAS;
(d) J. K. Schnobrich, O. Lebel, K. A. Cychosz, A. Dailly, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2010, 132, 13941 CrossRef CAS PubMed;
(e) J.-S. Hu, Y.-J. Shang, X.-Q. Yao, L. Qin, Y.-Z. Li, Z.-J. Guo, H.-G. Zheng and Z.-L. Xue, Cryst. Growth Des., 2010, 10, 4135 CrossRef CAS;
(f) R. P. Davies, R. Less, P. D. Lickiss, K. Robertson and A. J. P. White, Cryst. Growth Des., 2010, 10, 4571 CrossRef CAS;
(g) S. Barman, H. Furukawa, O. Blacque, K. Venkatesan, O. M. Yaghi and H. Berke, Chem. Commun., 2010, 46, 7981 RSC;
(h) H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed;
(i) M. E. Braun, C. D. Steffek, J. Kim, P. G. Rasmussen and O. M. Yaghi, Chem. Commun., 2001, 2532 RSC;
(j) D. T. Vodak, M. E. Braun, J. Kim, M. Eddaoudi and O. M. Yaghi, Chem. Commun., 2001, 2534 RSC.
-
(a) P. Kanoo, G. Mostafa, R. Matsuda, S. Kitagawa and T. K. Maji, Chem. Commun., 2011, 47, 8106 RSC;
(b) M.-S. Liu, Q.-Y. Yu, Y.-P. Cai, C.-Y. Su, X.-M. Lin, X.-X. Zhou and J.-W. Cai, Cryst. Growth Des., 2008, 8, 4083 CrossRef CAS;
(c) M. Frisch and C. L. Cahill, Cryst. Growth Des., 2008, 8, 2921 CrossRef CAS;
(d) C.-D. Wu, P. Ayyappan, O. R. Evans and W. Lin, Cryst. Growth Des., 2007, 7, 1690 CrossRef CAS;
(e) S. K. Ghosh, G. Savitha and P. K. Bharadwaj, Inorg. Chem., 2004, 43, 5495 CrossRef CAS PubMed.
-
(a) C. S. Hawes and P. E. Kruger, RSC Adv., 2014, 4, 15770 RSC;
(b) X.-L. Qi, C. Zhang, B.-Y. Wang, W. Xue, C.-T. He, S.-Y. Liu, W.-X. Zhang and X.-M. Chen, CrystEngComm, 2013, 15, 9530 RSC;
(c) S. Barman, H. Furukawa, O. Blacque, K. Venkatesan, O. M. Yaghi, G.-X. Jinc and H. Berke, Chem. Commun., 2011, 47, 11882 RSC;
(d) F. Salles, G. Maurin, C. Serre, P. L. Llewellyn, C. Knöfel, H. J. Choi, Y. Filinchuk, L. Oliviero, A. Vimont, J. R. Long and G. Férey, J. Am. Chem. Soc., 2010, 132, 13782 CrossRef CAS PubMed;
(e) M. Casarin, C. Corvaja, C. D. Nicola, D. Falcomer, L. Franco, M. Monari, L. Pandolfo, C. Pettinari and F. Piccinelli, Inorg. Chem., 2005, 44, 6265 CrossRef CAS PubMed.
-
(a) Z. Xia, Q. Wei, Q. Yang, C. Qiao, S. Chen, G. Xie, G. Zhang, C. Zhou and S. Gao, CrystEngComm, 2013, 15, 86 RSC;
(b) J.-Y. Wua, C.-W. Yang, H.-F. Chen, Y.-C. Jao, S.-M. Huang, C. Tsai, T.-W. Tseng, G.-H. Lee, S.-M. Peng and K.-L. Lu, J. Solid State Chem., 2011, 184, 1740 CrossRef PubMed;
(c) Y. G. Sun, X. Song, L. Wang, W. Yu, Y. Q. Wang, G. Xiong, M. Y. Guo and E. J. Gao, Russ. J. Coord. Chem., 2011, 37, 316 CrossRef CAS;
(d) Y.-Q. Sun, J. Zhang and G.-Y. Yang, Chem. Commun., 2006, 1947 RSC.
-
(a) M. H. Alkordi, J. A. Brant, L. Wojtas, V. Ch. Kravtsov, A. J. Cairns and M. Eddaoudi, J. Am. Chem. Soc., 2009, 131, 17753 CrossRef CAS PubMed;
(b) X.-F. Lin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m2039 CAS;
(c) S. Gao, C.-S. Gu, J. Y. Lu and Z. Ge, Inorg. Chim. Acta, 2005, 358, 828 CrossRef PubMed;
(d) L.-H. Huo, H. Zhao and J.-G. Zhao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m1672 Search PubMed.
-
(a) Z.-G. Gu, H.-C. Fang, P.-Y. Yin, L. Tong, Y. Ying, S.-J. Hu, W.-S. Li and Y.-P. Cai, Cryst. Growth Des., 2011, 11, 2220 CrossRef CAS;
(b) S.-Q. Zhang, F.-L. Jiang, M.-Y. Wu, R. Feng, J. Ma, W.-T. Xu and M.-C. Hong, Inorg. Chem. Commun., 2011, 14, 1400 CrossRef CAS PubMed;
(c) W. Wang, X. Niu, Y. Gao, Y. Zhu, G. Li, H. Lu and M. Tang, Cryst. Growth Des., 2010, 10, 4050 CrossRef CAS;
(d) W.-G. Lu, L. Jiang and T.-B. Lu, Cryst. Growth Des., 2010, 10, 4310 CrossRef CAS;
(e) X. Jing, H. Meng, G. Li, Y. Yu, Q. Huo, M. Eddaoudi and Y. Liu, Cryst. Growth Des., 2010, 10, 3489 CrossRef CAS.
-
(a) X. Li, B.-L. Wu, C.-Y. Niu, Y.-Y. Niu and H.-Y. Zhang, Cryst. Growth Des., 2009, 9, 3423 CrossRef CAS;
(b) W.-G. Lu, L. Jiang, X.-L. Feng and T.-B. Lu, Cryst. Growth Des., 2008, 8, 986 CrossRef CAS;
(c) J.-D. Lin, J.-W. Cheng and S.-W. Du, Cryst. Growth Des., 2008, 8, 3345 CrossRef CAS;
(d) W. Liu, L. Ye, X. Liu, L. Yuan, X. Lu and J. Jiang, Inorg. Chem. Commun., 2008, 11, 1250 CrossRef CAS PubMed;
(e) R.-Q. Zhong, R.-Q. Zou and Q. Xu, Microporous Mesoporous Mater., 2007, 102, 122 CrossRef CAS PubMed;
(f) R.-Q. Fang and X.-M. Zhang, Inorg. Chem., 2006, 45, 4801 CrossRef CAS PubMed;
(g) Y.-Q. Sun, J. Zhang and G.-Y. Yang, Chem. Commun., 2006, 4700 RSC;
(h) W.-G. Lu, L. Jiang, X.-L. Feng and T.-B. Lu, Cryst. Growth Des., 2006, 6, 564 CrossRef CAS;
(i) P. Mahata and S. Natarajan, Eur. J. Inorg. Chem., 2005, 2156 CrossRef CAS;
(j) Y.-Q. Sun, J. Zhang, Y.-M. Chen and G.-Y. Yang, Angew. Chem., Int. Ed., 2005, 44, 5814 CrossRef CAS PubMed.
-
(a) S. Wang, T. Zhao, G. Li, L. Wojtas, Q. Huo, M. Eddaoudi and Y. Liu, J. Am. Chem. Soc., 2010, 132, 18038 CrossRef CAS PubMed;
(b) Y. Liu, V. C. Kravtsov and M. Eddaoudi, Angew. Chem., Int. Ed., 2008, 47, 8446 CrossRef CAS PubMed;
(c) J.-Z. Gu, W.-G. Lu, L. Jiang, H.-C. Zhou and T.-B. Lu, Inorg. Chem., 2007, 46, 5835 CrossRef CAS PubMed;
(d) T. K. Maji, G. Mostafa, H.-C. Changa and S. Kitagawa, Chem. Commun., 2005, 2436 RSC;
(e) F. Zhang, Z. Li, T. Ge, H. Yao, G. Li, H. Lu and Y. Zhu, Inorg. Chem., 2010, 49, 3776 CrossRef CAS PubMed;
(f) X. Li, B. Wu, R. Wang, H. Zhang, C. Niu, Y. Niu and H. Hou, Inorg. Chem., 2010, 49, 2600 CrossRef CAS PubMed;
(g) Q. Xu, R.-Q. Zou, R.-Q. Zhong, C. Kachi-Terajima and S. Takamizawa, Cryst. Growth Des., 2008, 8, 2458 CrossRef CAS;
(h) M.-B. Zhang, Y.-M. Chen, S.-T. Zheng and G.-Y. Yang, Eur. J. Inorg. Chem., 2006, 1423 CrossRef CAS;
(i) C.-J. Li, S. Hu, W. Li, C.-K. Lam, Y.-Z. Zheng and M.-L. Tong, Eur. J. Inorg. Chem., 2006, 1931 CrossRef CAS;
(j) Y.-L. Wang, D.-Q. Yuan, W.-H. Bi, X. Li, X.-J. Li, F. Li and R. Cao, Cryst. Growth Des., 2005, 5, 1849 CrossRef CAS;
(k) W.-G. Lu, J.-Z. Gu, L. Jiang, M.-Y. Tan and T.-B. Lu, Cryst. Growth Des., 2005, 8, 192 CrossRef;
(l) X. Zhang, D. Huang, F. Chen, C. Chen and Q. Liu, Inorg. Chem. Commun., 2004, 7, 662 CrossRef CAS PubMed;
(m) M. H. Alkordi, Y. Liu, R. W. Larsen, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 12639 CrossRef CAS PubMed;
(n) R.-Q. Zou, H. Sakurai and Q. Xu, Angew. Chem., Int. Ed., 2006, 45, 2542 CrossRef CAS PubMed;
(o) W.-G. Lu, L. Jiang, X.-L. Feng and T.-B. Lu, Inorg. Chem., 2009, 48, 6997 CrossRef CAS PubMed;
(p) Y. Liu, V. C. Kravtsov, R. Larsen and M. Eddaoudi, Chem. Commun., 2006, 1488 RSC;
(q) W.-G. Lu, C.-Y. Su, T.-B. Lu, L. Jiang and J.-M. Chen, J. Am. Chem. Soc., 2006, 128, 34 CrossRef CAS PubMed.
-
(a) C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS PubMed;
(b) R. Kitaura, G. Onoyama, H. Sakamoto, R. Matsuda, S. Noro and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 2684 CrossRef CAS;
(c) Q.-H. Fan, Y.-M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385 CrossRef CAS PubMed.
-
(a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef CAS PubMed;
(b) W. A. Hermann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef;
(c) ed. F. Glorious, N-Heterocyclic carbenes in transition metal catalysis, Topics in Organometallic Chemistry, Springer-Verlag, Berlin/Heidelberg, 2007, vol. 21 Search PubMed.
- J. Yoon, S. K. Kim, N. J. Singh and K. S. Kim, Chem. Soc. Rev., 2006, 35, 355 RSC.
-
(a) J. M. Roberts, O. K. Farha, A. A. Sarjeant, J. T. Hupp and K. A. Scheidt, Cryst. Growth Des., 2011, 11, 4747 CrossRef CAS;
(b) K. Oisaki, Q. Li, H. Furukawa, A. U. Czaja and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 9262 CrossRef CAS PubMed;
(c) R. S. Crees, M. L. Cole, L. R. Hanton and C. J. Sumby, Inorg. Chem., 2010, 49, 1712 CrossRef CAS PubMed;
(d) X.-C. Chai, Y.-Q. Sun, R. Lei, Y.-P. Chen, S. Zhang, Y.-N. Cao and H.-H. Zhang, Cryst. Growth Des., 2010, 10, 658 CrossRef CAS;
(e) J. Chun, I. G. Jung, H. J. Kim, M. Park, M. S. Lah and S. U. Son, Inorg. Chem., 2009, 48, 6353 CrossRef CAS PubMed;
(f) L. Liu, Z. Li, B. Wang, G. Li, L. Wang, X. Meng and Z. He, Cryst. Growth Des., 2009, 9, 5244 CrossRef CAS;
(g) L. Han, S. Zhang, Y. Wang, X. Yan and X. Lu, Inorg. Chem., 2009, 48, 786 CrossRef CAS PubMed;
(h) Z. Fei, T. J. Geldbach, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2006, 45, 6331 CrossRef CAS PubMed;
(i) Z. Fei, T. J. Geldbach, D. Zhao, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2005, 44, 5200 CrossRef CAS PubMed.
- V. Karthik, I. A. Bhat and G. Anantharaman, Organometallics, 2013, 32, 7006 CrossRef CAS.
- C. Mohapatra, S. Tripathi, G. Anantharaman and V. Chandrasekhar, Cryst. Growth Des., 2014, 14, 3182 CAS.
-
(a) M. P. Hay, W. R. Wilson and W. A. Denny, Tetrahedron, 2000, 56, 645 CrossRef CAS;
(b) K. Hara, Y. Kanamori and M. Sawamura, Bull. Chem. Soc. Jpn., 2006, 79, 1781 CrossRef CAS.
-
G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Solution and Refinement; University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
-
V. A. Blatov, Multipurpose crystallochemical analysis with the program package TOPOS, IUCr CompComm Newsletter, 2006, vol. 7, p. 4, http://www.topos.ssu.samara.ru Search PubMed.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 22 CrossRef.
- R. Koner and I. Goldberg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, m37 CAS.
-
(a) G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565 CrossRef CAS PubMed;
(b) D. Braga, F. Grepioni and G. R. Desiraju, J. Organomet. Chem., 1997, 548, 33 CrossRef CAS;
(c) G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441 CrossRef CAS PubMed;
(d) G. R. Desiraju, Acc. Chem. Res., 1991, 24, 290 CrossRef CAS.
-
(a) S. P. Thomas, M. S. Pavan and T. N. G. Row, Chem. Commun., 2014, 50, 49 RSC;
(b) G. Bhattacharjya, G. Savithaa and G. Ramanathan, CrystEngComm, 2004, 6, 233 RSC.
- R. Alizadeh, K. Kalateh, Z. Khoshtarkib, R. Ahmadi and V. Amani, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1439 CAS.
-
(a) J. Heine and K. Müller-Buschbaum, Chem. Soc. Rev., 2013, 42, 9232 RSC;
(b) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed;
(c) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC.
- C. Banglin, W. Liangbo, Z. Fatima, Q. Guodong and E. B. Lobkovsky, J. Am. Chem. Soc., 2008, 130, 6718 CrossRef PubMed.
- P. Chakraborty, S. Mondal, S. Das, A. D. Jana and D. Das, Polyhedron, 2014, 70, 11 CrossRef CAS PubMed.
Footnote |
| † Additional figures, TGA, powder X-ray diffraction patterns. X-ray crystallographic data in CIF format have been deposited with the Cambridge Structural Database. CCDC 1027616–1027621 contains the supplementary crystallographic data for this paper. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce02215g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence