
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating the elusive crystal structure of 2,4,6-tris(2-pyrimidyl)-1,3,5-triazine†
Damir A.
Safin
a,
Nikolay A.
Tumanov
b,
Alicea A.
Leitch
a,
Jaclyn L.
Brusso
a,
Yaroslav
Filinchuk
*b and
Muralee
Murugesu
*a
aDepartment of Chemistry, University of Ottawa, 10 Marie Curie Private, Ottawa, ON, Canada K1N 6N5. E-mail: m.murugesu@uottawa.ca; Fax: +1 (613) 562 5170; Tel: +1 (613) 562 5800 ext. 2733
bInstitute of Condensed Matter and Nanosciences, Université catholique de Louvain, Place L. Pasteur 1, 1348 Louvain-la-Neuve, Belgium. E-mail: yaroslav.filinchuk@uclouvain.be
First published on 4th February 2015
Abstract
2,4,6-Tris(2-pyrimidyl)-1,3,5-triazine (TPymT) has been synthesized using two approaches: via trimerization of 2-cyanopyrimidine at 160 °C and by the reaction of 2-cyanopyrimidine with pyrimidine-2-carboximidamide. It was found that the two different synthetic pathways to TPymT yielded either a single polymorph or a mixture of two polymorphs; both of which were studied for the first time by X-ray powder diffraction (XRPD) and synchrotron X-ray powder diffraction (SXRPD). The crystal structure of the polymorph α-TPymT was determined by single crystal X-ray diffraction (SCXRD). Crystal structures of 2-cyanopyrimidine and pyrimidine-2-carboximidamide were also elucidated by SXRPD.
2,4,6-Tris(2-pyrimidyl)-1,3,5-triazine (TPymT) has been known for about 55 years, when Case and Koft isolated the product with a very low yield from 2-cyanopyrimidine on standing for several months.1 About 20 years later, Lerner and Lippard described the synthesis of TPymT from the same precursor with an extremely high yield.2 Although TPymT is very attractive as a potential ligand for metal complexation due to the presence of three fused terpyridine-like chelating sites, to the best of our knowledge, only a few works have been reported on the coordination chemistry of TPymT.3 In fact, until recently only two reported crystal structures involving coordination of TPymT to PbII had been published.3a,b This can be attributed to the sensitivity of the triazine fragment towards hydrolysis in the presence of metal cations under mild conditions.2,4 As such, the coordination chemistry of TPymT remains an on going challenge. With this in mind we have recently directed our attention to the synthesis and study of TPymT-based coordination compounds and, as a first step, we reported the unprecedented dinuclear CdII complex, [Cd2(TPymT)(H2O)6(SO4)2]·H2O.5 Furthermore, we have reported a pseudo-polymorph [Pb2(TPymT)(NO3)4]n (ref. 6) of a previously published lead structure.3a These polynuclear complexes show a glimpse of great potential of TPymT as a multidentate ligand. To prevent TPymT degradation through hydrolysis and subsequently master the coordination chemistry, we need to understand the stability of TPymT on its own.
Surprisingly, the crystal structure of TPymT is still unknown, although its close analogues 2,4,6-tris(2-pyridyl)-1,3,5-triazine and 2,4,6-tris(4-pyridyl)-1,3,5-triazine have been characterized by single crystal X-ray diffraction.7 This is likely due to the extremely poor solubility of TPymT in common solvents. Moreover, the crystal structure of the precursor, 2-cyanopyridine, was also unknown. This may be explained by the formation of micro-crystals, which are not suitable for single crystal X-ray diffraction. To that end, we herein report the crystal structures of 2-cyanopyrimidine and two polymorphs of TPymT, which were established through X-ray powder diffraction (XRPD) and synchrotron X-ray powder diffraction (SXRPD). The crystal structure of the main polymorph of TPymT was determined by single crystal X-ray diffraction (SCXRD). Additionally, we report a novel and more efficient synthetic route for the preparation of TPymT, along with SXRPD analysis of the intermediate, pyrimidine-2-carboximidamide.
TPymT can be obtained via two different synthetic approaches as outlined in Scheme 1. The first involves trimerization of 2-cyanopyrimidine at 160 °C5 affording TPymT in 17% yield. Alternatively, TPymT can be prepared by heating 2-cyanopyrimidine under pressure in the presence of pyrimidine-2-carboximidamide, which was synthesized by heating a solution of 2-cyanopyrimidine and ammonia under pressure for several days. While TPymT can be prepared following both synthetic sequences, the latter approach resulted in significantly higher yields (60% vs. 17%) and cleaner crude material. We therefore suggest this finding as a new highly efficient synthetic approach for TPymT. Interestingly, the first synthetic route (i.e., trimerization of 2-cyanopyrimidine) leads to the formation of a single polymorph α-TPymT, whereas the second pathway affords a mixture of two polymorphs α-TPymT and β-TPymT.
 | ||
| Scheme 1 Preparation of α-TPymT and β-TPymT. | ||
The isolated compounds were initially characterized by FTIR and NMR spectroscopy. The FTIR spectra of 2-cyanopyrimidine, pyrimidine-2-carboximidamide and TPymT each contain an intense band for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretches centred at about 1580 cm−1 (Fig. 1). The aromatic C–H groups in the spectra of the latter two compounds were found as a band at 3060 cm−1, while the same stretches as well as a band for the C
N stretches centred at about 1580 cm−1 (Fig. 1). The aromatic C–H groups in the spectra of the latter two compounds were found as a band at 3060 cm−1, while the same stretches as well as a band for the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group in the spectrum of 2-cyanopyrimidine are too weak to be clearly observed. The spectrum of pyrimidine-2-carboximidamide also exhibits a set of intense bands for the NH and NH2 groups at 1660, 3110, 3290 and 3412 cm−1.
N group in the spectrum of 2-cyanopyrimidine are too weak to be clearly observed. The spectrum of pyrimidine-2-carboximidamide also exhibits a set of intense bands for the NH and NH2 groups at 1660, 3110, 3290 and 3412 cm−1.
 | ||
| Fig. 1 FTIR spectra of 2-cyanopyrimidine (black), pyrimidine-2-carboximidamide (red) and TPymT (blue). | ||
The 1H NMR spectra of 2-cyanopyrimidine, pyrimidine-2-carboximidamide and TPymT in DMSO-d6 each exhibit peaks for the pyrimidine fragment at 7.63–7.87 and 8.95–9.16 ppm. The spectrum of pyrimidine-2-carboximidamide also contains a broad singlet peak for the NH and NH2 protons at 7.12 ppm. The 13C NMR spectra of 2-cyanopyrimidine and pyrimidine-2-carboximidamide each contain four peaks. Due to the low solubility of TPymT in organic solvents, 13C NMR was not possible.
To shed light on the electronic properties of 2-cyanopyrimidine, pyrimidine-2-carboximidamide and TPymT, diffuse reflectance spectra were recorded (Fig. 2). These spectra exhibit a broad band with several maxima in the UV region corresponding to intra-ligand transitions. The spectra of pyrimidine-2-carboximidamide and TPymT additionally contain a shoulder in the visible range at 375–600 nm, which is responsible for the observed beige colour of these compounds.
 | ||
| Fig. 2 Normalized Kubelka-Munk spectra of 2-cyanopyrimidine (black), pyrimidine-2-carboximidamide (red) and TPymT (blue). | ||
In order to elucidate the molecular structures of 2-cyanopyrimidine and pyrimidine-2-carboximidamide as well as their spatial arrangement in the crystalline lattice, synchrotron radiation powder X-ray studies were conducted (Fig. 3, Tables S1 and S2 in ESI†). Obtained powder patterns were solved in the orthorhombic P212121 (2-cyanopyrimidine) and triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (pyrimidine-2-carboximidamide) space groups. The asymmetric units of 2-cyanopyrimidine and pyrimidine-2-carboximidamide each contain one molecule.
(pyrimidine-2-carboximidamide) space groups. The asymmetric units of 2-cyanopyrimidine and pyrimidine-2-carboximidamide each contain one molecule.
 | ||
| Fig. 3 Rietveld refinement plots (λ = 0.68884 Å) of 2-cyanopyrimidine (top) and pyrimidine-2-carboximidamide (bottom). Experimental and calculated data are shown in red symbols and black line, respectively. Blue line shows the difference curve, while green marks show Bragg positions. Insets show molecular structures of 2-cyanopyrimidine and pyrimidine-2-carboximidamide. | ||
With respect to the packing of 2-cyanopyrimidine, the molecules form zigzag-like layers (Fig. 4). Molecules of pyrimidine-2-carboximidamide form parallel ribbons along the b axis via intermolecular N–H⋯N hydrogen bonds (Fig. 5, Table S3 in ESI†). Positions of the hydrogen atoms, determined from XRPD, are often inaccurate; however, based on the positions of non-hydrogen atoms we assume that the CNH hydrogen atom is not involved in the hydrogen bond formation due to the lack of neighbouring acceptor atoms. The ribbons are further linked through weak π⋯π stacking interactions (Table S4 in ESI†).
 | ||
| Fig. 4 Zigzag-like layers in the crystal packing of 2-cyanopyrimidine along the a (top) and b (bottom) axes. | ||
 | ||
| Fig. 5 A ribbon-like structure, formed due to intermolecular hydrogen bonds, in the crystal packing of pyrimidine-2-carboximidamide along the b axis. | ||
The crystal structure of α-TPymT was initially solved using ab initio from XRPD data of the sample obtained via the trimerization reaction using direct space Monte Carlo methods and refined by the Rietveld method. The experimental powder pattern was indexed easily and unambiguously due to the presence of one phase in the sample. However, choice of the space group remained ambiguous since P31, P61 and P3121 space groups have a similar fit in the Le Bail refinement. Numerous attempts were therefore taken to solve the structure in these three space groups and the best results for each space group have been selected. The crystal structure refinement was further complicated by the preferred orientation; nevertheless, the final structure from XRPD data was refined in the P31 space group (for additional discussion about the choice of a space group see ESI†).
In order to confirm this assignment, single crystal X-ray analysis was required. After numerous attempts, crystals of α-TPymT suitable for SCXRD were produced directly from the trimerization of 2-cyanopyrimidine. However, even the best selected crystal only diffracted up to 1.0 Å−1. Thus the structural model determined from XRPD was used as the initial starting point in the single-crystal data refinement. This model, in the P31 space group, was tested for any missing symmetry elements in PLATON8 and a two-fold axis was found. The space group was therefore changed to P3121 and applied to the final structure refinement.
The asymmetric unit of α-TPymT contains two independent halves of two molecules (Fig. 6, Table S5 in ESI†) and the two-fold axis generates the remaining halves to complete the molecules. The crystal structure consists of alternating flat layers, packed orthogonal to the c axis. The torsion angles between the planes, formed by the triazine and pyrimidine rings, are −24(4), −24(4) and 36.2(14)° in one molecule and −37(4), 21.8(14) and 21.8(14)° in the other.
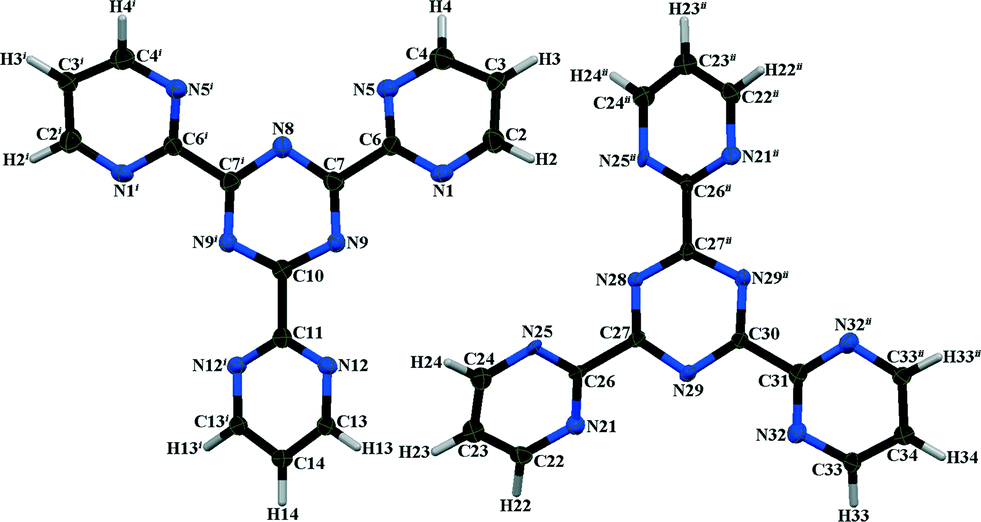 | ||
| Fig. 6 Molecular structure of two independent molecules of α-TPymT. Symmetry codes: i: 1 + x − y, 2 − y, 2/3 − z; ii: −x, −x + y, 1/3 − z. | ||
The two symmetry independent molecules in α-TPymT each produce two sets of layers; however, they are very similar and only differ slightly with respect to the orientation of the pyrimidine rings relative to the triazine ring (Fig. 7). The interlayer distances are approximately 3.53 and 3.63 Å, which is slightly offset (±1.3%) from that of c/6 = 3.578 Å. Such a layered structure leads to the appearance of a preferred orientation in the experimental XRPD.
 | ||
| Fig. 7 Crystal packing of α-TPymT along the b axis (H-atoms were omitted for clarity). Symmetry independent molecules are shown in different colours. | ||
Before obtaining single crystals of TPymT, we tried to determine a more detailed structure of TPymT using SXRPD data, which was collected from a sample obtained by the reaction of 2-cyanopyrimidine with pyrimidine-2-carboximidamide. From this sample, the diffraction pattern revealed the same peaks as were observed for the α-TPymT polymorph as well as several additional peaks that did not match the α-TPymT unit cell. These peaks were successfully indexed in the monoclinic system (Fig. 8), suggesting that the second monoclinic phase corresponds to another polymorph β-TPymT. This assumption is based on the following arguments: i) if the unit cell of the second phase contains two independent molecules, then its volume per molecule is very close to that of α-TPymT (341.16 and 334.08 Å3 for α- and β-polymorphs, respectively); ii) some of the unit cell parameters are similar, indicating the similarity between these phases; iii) comparison of the XRPD patterns of the two phases shows that the peaks of the second phase β-TPymT do not correspond to the starting materials 2-cyanopyrimidine and pyrimidine-2-carboximidamide (Fig. 3) or other possible impurities, which was further confirmed by IR and NMR data.
 | ||
| Fig. 8 Le Bail fit to the synchrotron radiation X-ray powder pattern (λ = 0.821693 Å) of the sample obtained by the reaction of 2-cyanopyrimidine with pyrimidine-2-carboximidamide. Experimental and calculated data are shown in red symbols and black line, respectively. Blue line shows the difference curve, while green marks show Bragg positions for α-TPymT (top row) and β-TPymT (bottom row). | ||
Analysis of systematic absences of reflections has shown that the structure of β-TPymT does not contain any glide planes, limiting a choice of the possible space group to P2, P21, Pm, P2/m and P21/m. Furthermore, unless TPymT is flat, a mirror plane in the structure is unlikely to be compatible with the molecular symmetry. This therefore narrows the choice of the space group for β-TPymT to P2 or P21. Based on the reflection intensities of the two phases, the content of β-TPymT can be roughly estimated to ~10%.
We assume that the sample obtained by reacting 2-cyanopyrimidine with pyrimidine-2-carboximidamide is an example of polytypism, which is a special case of polymorphism of layered structures when the two-dimensional translations within the layers are essentially preserved. Each polymorph of this type can be regarded as built up by stacking layers of a nearly identical structure, and they differ only in their stacking sequence. In our case, TPymT molecules form such layers in α-TPymT (Fig. 7) and, based on the unit cell parameters, also in β-TPymT. In both polymorphs, the molecules of TPymT are almost parallel to the ac plane. This might result in close values of interplanar spacings and lead to a significant overlap of diffraction peaks from the two phases. Such an overlap, accompanied with the preferred orientation, makes it impossible to extract intensities of peaks for two crystalline phases and, hence, to solve the structure of β-TPymT from SXRPD data.
In summary, for the first time we have obtained the crystal structure of TPymT and determined that it crystallizes in the trigonal P3121 space group to produce the polymorph α-TPymT. It was also established that the polymorph α-TPymT is the only product formed during the trimerization reaction of 2-cyanopyrimidine, while the reaction of 2-cyanopyrimidine with pyrimidine-2-carboximidamide leads to a mixture of α-TPymT and a second polymorph β-TPymT. Crystal structures of the precursors 2-cyanopyrimidine and pyrimidine-2-carboximidamide were also elucidated by SXRPD. These results are critical to understanding the supramolecular arrangement and packing of TPymT. Furthermore, the new, more efficient synthetic route for the isolation of TPymT opens an important avenue for large scale synthesis as well as its use in coordination chemistry as a counterpart for terpy or pyrimidine ligand systems.
Experimental
Physical measurements
Infrared spectra were recorded with a Varian 640 FTIR spectrometer equipped with an ATR in the 500–4000 cm−1 range. 1H and 13C NMR spectra in DMSO-d6 were obtained on a Bruker Avance 300 MHz spectrometer at 25 °C. Diffuse reflectance spectra were obtained with a Varian Cary 100 spectrometer using polytetrafluoroethylene (PTFE) as a reference. Kubelka-Munk spectra were normalized to allow meaningful comparisons.2-Cyanopyrimidine
M. p.: 40–44 °C. 1H NMR, δ: 7.87 (t, 3JH,H = 5.0 Hz, 1H, p-CH, pyrimidine), 9.02 (d, 3JH,H = 5.0 Hz, 2H, m-CH, pyrimidine) ppm. 13C NMR, δ: 115.91, 124.76, 143.94, 158.72 ppm.Synthesis of pyrimidine-2-carboximidamide
In a glass pressure vessel, ammonia gas (Linde) was bubbled through a solution of 2-cyanopyrimidine (95.1 mmol, 10.0 g; Oakwood Chemicals) in 100 mL MeCN (distilled over P2O5, stored on 4 Å molecular sieves) cooled on an ice bath for approximately 30 min. The flask was then sealed and heated at 110 °C for 3 days. The resulting red solution was cooled to room temperature, the vessel was carefully vented, and the brown crystalline solid of pyrimidine-2-carboximidamide was filtered off and washed with MeCN. Crude yield: 9.10 g (78%). Recrystallization from MeCN afforded pale beige needles. M. p.: 159–163 °C. 1H NMR, δ: 7.12 (br. s, 3H, NH + NH2), 7.63 (t, 3JH,H = 4.9 Hz, 1H, p-CH, pyrimidine), 8.95 (d, 3JH,H = 4.9 Hz, 2H, m-CH, pyrimidine) ppm. 13C NMR, δ: 121.81, 156.16, 157.52, 159.64 ppm. Anal. calcd. for C5H6N4: C, 49.17; H,4.95; N, 45.88. Found: C, 49.02; H, 4.90; N, 45.60.Synthesis of α-TPymT
α-TPymT was obtained using the synthetic procedure described by Lerner and Lippard;2 however, the yield was much lower than reported regardless of the reaction time. 2-Cyanopyrimidine (52 mmol, 5.5 g) was heated with stirring in a stoppered flask at 160 °C for 72 h. The resulting product was washed with diethyl ether (5 × 20 mL) to remove unreacted starting material. Yield: 0.96 g (17%). M. p.: >350 °C. 1H NMR, δ: 7.82 (t, 3JH,H = 4.8 Hz, 3H, p-CH, pyrimidine), 9.16 (d, 3JH,H = 4.8 Hz, 6H, m-CH, pyrimidine) ppm.Synthesis of α-TPymT and β-TPymT
To a glass pressure vessel under a blanket of N2(g), pyrimidine-2-carboximidamide (37.8 mmol, 4.62 g), 2-cyanopyrimidine (75.5 mmol, 7.93 g) and 100 mL MeCN (distilled over P2O5, stored on 4 Å molecular sieves) were loaded. The vessel was sealed and heated at 110 °C for 5 days. The green-brown slurry was cooled to room temperature and filtered. The crude solid was washed in hot EtOAc to afford a beige powder. Yield: 7.17 g (60%).X-ray powder diffraction (XRPD)
The XRPD pattern for the bulk sample of α-TPymT was measured using a Rigaku Ultima IV X-ray powder diffractometer (Cu Kα radiation, graphite monochromator, scintillation counter). The parallel beam mode was used to collect the data. Powder pattern was indexed in trigonal/hexagonal crystal system using the DICVOL04 program.9 Crystal structure was solved using direct space methods with the FOX program10 and refined by the Rietveld method using the Fullprof suite.11 Initial molecular model was constructed in the Mercury 3.0 program,12 using the 2,4,6-tris(4-pyridyl)-1,3,5-triazine molecule13 as a starting model, which was modifed. The preferred orientation, using the March–Dollase model for the (100) direction, and antibump restraints for the H⋯H distances (d(H⋯H) > 2.25 Å), was introduced at the final stage of the refinement.Single-crystal X-ray diffraction (SCXRD)
Inhouse data for α-TPymT were collected on a MAR345 image plate using Mo Kα radiation focused by a Xenocs Fox3D mirror at 150 K. Few crystals were tested and the best available dataset were chosen. The data were integrated using the CrysAlisPro14 software and the multi-scan absorption correction was applied. Data was cut off up to 1.0 Å−1. The structural model was refined by full-matrix least squares on |F|2 using SHELXL-2014 (ref. 15) and the shelXLe shell.16 Relative restrains on the C–C distances were used to make all length of bonds between pyrimidine and triazine ring simular to each other. All non-hydrogen atoms were refined anisotropically, but restrains were applied on thermal parameters. Hydrogen atoms were placed on calculated positions in riding mode with temperature factors fixed at 1.2 times Ueq of the parent atoms. Two-component inversion twinning was introduced at the final stage of the refinement.Synchrotron X-ray powder diffraction (SXRPD)
The SXRPD patterns for the bulk sample of a mixture of α-TPymT and β-TPymT, 2-cyanopyrimidine and pyrimidine-2-carboximidamide were measured on the Swiss-Norwegian beamline BM1A at the European Synchrotron Radiation Facility (transmittion geometry, PILATUS 2M pixel detector, λ = 0.821693 Å for a mixture of α-TPymT and β-TPymT, and λ = 0.68884 Å for 2-cyanopyrimidine and pyrimidine-2-carboximidamide). Powder patterns were indexed using the FOX program.10 Crystal structures were solved using direct space methods with the FOX program10 and refined by the Rietveld method using the Fullprof suite.11 Initial molecular models were constructed in the Mercury 3.0 program,12 using the corresponding molecular fragment based on the single-crystal structural models taken from the Cambridge Structural Database (QOFBUJ17 for 2-cyanopyrimidine and CONYAF10 (ref. 18) for pyrimidine-2-carboximidamide). The preferred orientation in the March–Dollase model for the (011) direction was introduced at the final stage of the structure determination for 2-cyanopyrimidine., a = 5.55795(14), b = 7.25941(12), c = 7.7260(2) Å, α = 84.431(2), β = 71.913(3), γ = 80.163(2)°, V = 291.643(12) Å3, Z = 2, ρ = 1.391 g cm−3, μ(λ = 0.68884 Å) = 0.089 mm−1, Rp = 0.0639, Rwp = 0.0683, RI = 0.1630.
Acknowledgements
This work was financially supported by the NSERC-DG, CFI, ORF, and ERA. We acknowledge the Fonds Spéciaux de Recherche (UCL) for the incoming postdoctoral fellowship co-funded by the Marie Curie actions of the European Commission granted to N. A. Tumanov. We thank ESRF for the beamtime allocation at the SNBL.Notes and references
- F. H. Case and E. Koft, J. Am. Chem. Soc., 1959, 81, 905 CrossRef CAS.
- E. I. Lerner and S. J. Lippard, J. Am. Chem. Soc., 1976, 98, 5397 CrossRef CAS.
- (a) E. I. Lerner and S. J. Lippard, Inorg. Chem., 1977, 16, 1537 CrossRef CAS; (b) A. M. Garcia, D. M. Bassani, J.-M. Lehn, G. Baum and D. Fenske, Chem. – Eur. J., 1999, 5, 1234 CrossRef CAS; (c) C. Metcalfe, S. Spey, H. Adams and J. A. Thomas, J. Chem. Soc., Dalton Trans., 2002, 4732 RSC; (d) C. Metcalfe, C. Rajput and J. A. Thomas, J. Inorg. Biochem., 2006, 100, 1314 CrossRef CAS PubMed; (e) E. Jakubikova, R. L. Martin and E. R. Batista, Inorg. Chem., 2010, 49, 2975 CrossRef CAS PubMed.
- (a) E. I. Lerner and S. J. Lippard, Inorg. Chem., 1977, 16, 1546 CrossRef CAS; (b) D. Cangussu de Castro Gomes, H. O. Stumpf, F. Lloret, M. Julve, V. González, H. Adams and J. A. Thomas, Inorg. Chim. Acta, 2005, 358, 1113 CrossRef PubMed; (c) T. Glaser, H. Theil, I. Liratzis, T. Weyhermüller and E. Bill, Inorg. Chem., 2006, 45, 4889 CrossRef CAS PubMed.
- D. A. Safin, Y. Xu, I. Korobkov, D. L. Bryce and M. Murugesu, CrystEngComm, 2013, 15, 10419 RSC.
- D. A. Safin, K. M. N. Burgess, I. Korobkov, D. L. Bryce and M. Murugesu, CrystEngComm, 2014, 16, 3466 RSC.
- (a) M. G. B. Drew, M. J. Hudson, P. B. Iveson, M. L. Russell and C. Madic, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 985 Search PubMed; (b) J. Janczak, M. Sledz and R. Kubiak, J. Mol. Struct., 2003, 659, 71 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- A. Boultif and D. Louër, J. Appl. Crystallogr., 2004, 37, 724 CrossRef CAS.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55 CrossRef.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef PubMed.
- J. Janczak, M. Śledź and R. Kubiak, J. Mol. Struct., 2003, 71, 659 Search PubMed.
- Agilent Technologies. CrysAlis PRO, 2013 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
- Y. V. Kokunov and Y. E. Gorbunova, Russ. J. Inorg. Chem., 2007, 52, 1530 CrossRef.
- A. Marsura, C. L. Duc and G. Gellon, Tetrahedron Lett., 1984, 25, 4509 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2, Tables S1–S5. CCDC reference number 975002 (α-TPymT, the structure refined from the XRPD data), 1036571 (α-TPymT, the structure refined from the SCXRD data), 1014033 (2-cyanopyrimidine) and 1014032 (pyrimidine-2-carboximidamide). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce00097a |
| This journal is © The Royal Society of Chemistry 2015 |