
Structural characterization of LASSBio-1289: a new vasoactive N-methyl-N-acylhydrazone derivative†
Juliana Alves Pereira
Sato
a,
Fanny Nascimento
Costa
a,
Miguel Divino
da Rocha
bc,
Eliezer J.
Barreiro
bc,
Carlos Alberto Manssour
Fraga
bc,
Francesco
Punzo
d and
Fabio Furlan
Ferreira
 *a
*a
aCenter of Natural and Human Sciences (CCNH), Federal University of ABC (UFABC), Av. dos Estados, 5001, Santo André, SP 09210-580, Brazil. E-mail: fabio.furlan@ufabc.edu.br
bLASSBio, Institute of Biomedical Sciences, Federal University of Rio de Janeiro (UFRJ), Av. Carlos Chagas Filho, 373, Rio de Janeiro, RJ 21941-902, Brazil
cGraduate Program of Chemistry, Institute of Chemistry, Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil
dDipartimento di Scienze del Farmaco, Sezione Chimica, Università degli Studi di Catania, Catania 95125, Italy
First published on 21st October 2014
Abstract
LASSBio-1289 compound has been found to promote intense vasodilation and antihypertensive activity. It is an innovative compound, without structural similarities to the three main classes of calcium antagonists (1,4-dihydropyridines, benzothiazepines and phenylalkylamines) commonly used. A complete knowledge of the structure, including stereochemistry, is essential to lead optimization in drug discovery. For this reason, in this work we determined the crystal structure of this novel vasoactive N-methyl-N-acylhydrazone derivative by means of X-ray powder diffraction data and an ab initio simulating annealing approach, allowing us to observe the relative configuration E of the imine double bond and its conformation as well as its most relevant intermolecular interactions. These findings were also checked by a FTIR analysis and confirmed in solution by NMR. The compound crystallized under a monoclinic crystal system with space group P21/c and unit cell parameters a = 14.5118(3) Å, b = 12.1374(2) Å, c = 7.5498(1) Å, β = 91.113(1)°, V = 1329.53(4) Å3, Z = 4, Z′ = 1 and ρcalc = 1.44042(4) g cm−3. Moreover, a crystal morphology prediction, experimentally compared with SEM inferred images, allowed a direct comparison of the microcrystalline habit and quality, allowing a study of the potential solvent effect on the crystal growth.
Introduction
Hypertension or high-blood pressure is considered a multi-factorial disease being an important worldwide public-health challenge due to its high frequency and concomitant risks of cardiovascular as well as other diseases.1,2 It has been identified as the leading risk factor for mortality, being ranked as the third cause of disability-adjusted life-years.3 The worldwide prevalence of hypertension has been widely reported.4–7During the past century, hypertension has changed from a minor cause of death and disability to one of the major contributors to the global burden of disease.8
Kearney et al.9 indicated that more than a quarter of the world's adult population – totalizing nearly one billion people – had hypertension in 2000, and that this proportion will increase to 29% – 1.56 billions – by the year 2025. Prevention, detection, treatment, and control of this condition should receive high priority. These pieces of information underscore the importance of an effective health policy, medical care, and public health actions that improve hypertension prevention and control.10
Although several effective treatments for hypertension are available, novel therapies to reduce elevated blood pressure, improve blood-pressure control, treat resistant hypertension, and reduce the associated cardiovascular risk factors are still required.11 In this context, some bioactive N-acylhydrazone (NAH) derivatives from safrole, synthesized in LASSBio® – Laboratory of Evaluation and Synthesis of Bioactive Substances of the Federal University of Rio de Janeiro (UFRJ) – have been found to promote intense vasodilation and antihypertensive activity.12–14
LASSBio-294 has been described as a potent cardiac inotropic agent15 with vasodilatory properties (IC50 = 74 μM). Several derivatives of LASSBio-294 have been synthesized in an effort to identify new drug candidates with selective vasodilator activity. In this context, the LASSBio-785 synthesis was planned by introducing a methyl group linked to the amide nitrogen of the NAH moiety of LASSBio-294, thus showing a significant improvement in vasodilatory properties (IC50 = 10.2 μM), being seven times more potent than LASSBio-294.14,16,17 Considering the bioprofile of LASSBio-294, some synthetic analogues were obtained by changing the electronic density of the thienyl subunit and modifying the stereoelectronic behaviour of the acylhydrazone group through its N-alkylation. These analogues were evaluated in vascular smooth muscle, in an effort to identify new drug candidates with vasodilatory properties and fewer side effects. The results showed an important pharmacophoric profile for the thienyl ring.14 Thus, a new LASSBio-294 regioisomer was synthesized by the replacement of the 2-thienyl ring with a 3-thienyl ring, (3-thienylidene)-3,4-methylenedioxybenzoylhydrazide – LASSBio-897 – which increased its vasorelaxant activity (IC50 = 0.35 μM) as compared to the parent compound.18
The LASSBio-1289 compound, (E)-N-methyl-N′-(thiophen-3-yl-methylene) benzo[d][1,3]dioxole-5-carbohydrazide, was also designed from the parent compound LASSBio-294 through the use of medicinal chemistry strategy of bioisosteric replacement of aromatic rings.15,19 In addition, the N-methylation of the NAH group was used to improve the vasodilatory profile of the parent compound by inducing conformational changes (Fig. 1).16–18
 | ||
| Fig. 1 NAH derivatives of LASSBio-294, new drug candidates with vasodilator activity. | ||
After the planning and synthesis of LASSBio-1289, its cardiovascular properties were evaluated. Pereira et al.20 demonstrated that LASSBio-1289 induces both endothelium-independent vasorelaxation involving the inhibition of Ca2+ influx through L-type Ca2+ channels in aorta from Wistar–Kyoto (WKY) and spontaneously hypertensive rats (SHR), as well as endothelium-dependent relaxation mediated by the nitric oxide/cyclic GMP (Guanosine Monophosphate) pathway in WKY rats.
In a recent study, Pereira et al.21 also investigated the actions of LASSBio-1289 on monocrotaline (MCT)-induced pulmonary arterial hypertension (PAH) in rats. Oral treatment with LASSBio-1289 effectively decreased pulmonary artery diameter and right ventricle area, assessed by echocardiography. LASSBio-1289 was effective in attenuating MCT-induced PAH in rats, and its beneficial effects were likely mediated by the inhibition of extracellular Ca2+ influx through L-type voltage-gated Ca2+ channels in the pulmonary artery.21
LASSBio-1289 is an innovative compound, without any structural similarity to the three main classes of calcium antagonists (1,4-dihydropyridines, benzothiazepines and phenylalkylamines). This new NAH derivative was 25-fold more potent than the parent compound LASSBio-294 in promoting aorta relaxation. This increase in potency may be due to structural changes such as the N-methylation and isosteric replacement of the 2-thienyl by the 3-thienyl ring. In addition, LASSBio-1289 was 90-fold more potent than the precursor LASSBio-897 in aorta from SHR.20
Many factors are involved in the task of describing the relationship between chemical structure and pharmacological activity of a new drug prototype. Among these factors, the study of atomic-level structure can be included.
The aim of this work is to carry out the structural characterization of LASSBio-1289 in the solid state. This study can contribute to a better understanding of the full pharmacodynamic profiles and physicochemical properties of this class of compounds. Since many NAH derivatives, as LASSBio-1289, do not create crystals suitable for single crystal X-ray diffraction, the methodology of structure determination from X-ray powder diffraction data has been employed as a technique able to define the relative configuration of the compounds, which is directly related to their biological activity. This technique can be applied to characterize possible intermolecular and intramolecular interactions beyond providing information about conformational freedom, flatness as well as to distinguish the exact orientation of the structural groups of the molecule.22–26 FTIR was used to evaluate the possible bond deviation – both axial and angular – from the data reported in the literature for this class of compounds. The present analysis was also performed in solution by means of NMR. A crystal morphology prediction, compared with the experimentally inferred SEM images, was performed. This study, coupled with the above-mentioned analyses, allowed a description at the molecular level of the main forces involved in the crystal growth and its final habit. Furthermore, the potential role of solvents on crystal morphology has been investigated, thus providing also key information about the possible tableting of this active pharmaceutical ingredient.27,28 In this way, we also try to help shedding light on its physicochemical properties and its pharmacokinetiks characteristics.
Experimental
Material
The LASSBio-1289 compound was synthesized, in LASSBio®, following the procedure described in the literature.20 The sample was gently hand-grinded in an agate mortar in order to get a fine powder suitable for X-ray powder diffraction analysis.Sample purity
The sample purity (99%) was analysed by reversed-phase high-performance liquid chromatography (HPLC) experiment using a Shimadzu LC20AD apparatus (Shimadzu, Co., Kyoto, Japan), Kromasil 100-5 C18 (4.6 mm × 250 mm column and SPD-M20A detector (diode array)) (Kromasil, Bohus, Sweden). The chromatogram was obtained with a 20 μL sample injection, 1 mL min−1 constant flow rate (acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water – 60:40) and analysed at λ = 254 nm. Data were acquired by LC Solution software, version 4.0. The solvents (HPLC standard grade) were obtained from TEDIA®.
:water – 60:40) and analysed at λ = 254 nm. Data were acquired by LC Solution software, version 4.0. The solvents (HPLC standard grade) were obtained from TEDIA®.
Accurate mass was measured using a Bruker MicrOTOF II (Bruker Daltonik GmbH, Bremen, Germany) equipment. Ionization was carried out by an ESI source (not heated). We obtained a mass of m/z = 311.0460 which refers to a [M + Na+] ([M + Na+]calc. = 311.0460). The obtained spectrum is described in ESI.†
X-ray powder diffraction
X-ray powder diffraction data were collected at room temperature on a STADI-P powder diffractometer (Stoe®, Darmstadt, Germany) in transmission geometry by using a Kα1 (λ = 1.54056 Å) wavelength emitted by a Cu anode and selected by a curved Ge(111) crystal, with a tube voltage of 40 kV and a current of 40 mA. The sample was packed between two 0.014 mm thick cellulose-acetate foils in a sample holder that was held spinning during data collection. The diffracted intensities were collected by a silicon microstrip detector, Mythen 1K (Dectris®, Baden, Switzerland), in the range from 2° to 60°, with step sizes of 0.015° and 60 s of integration time at each 1.05°.Indexing
X-ray powder diffraction data were used for indexing the first 20 reflections of the pattern using the Topas-Academic v.5 software program.29 The region comprised by these reflections was fitted without corrections for the zero point of the diffractometer and the Lorentz-polarization factor, yielding the following values for a monoclinic space group (P21/c): a = 14.5062 Å, b = 12.1341 Å, c = 7.5483 Å, β = 91.113°, V = 1328.399 Å3 and Gof = 90.08.30 Then, a Pawley refinement31 was done to confirm the choice of space group as well as unit cell parameters. The values of unit cell parameters as well as R-factors32 and goodness-of-fit indicator found after the refinement were: a = 14.5108(4) Å, b = 12.1373(3) Å, c = 7.5499(1) Å, β = 91.110(1)°, V = 1329.45(6) Å3, Rwp = 3.836%, Rexp = 3.592% and χ2 = 1.068, resulting in a good visual fit of the whole pattern.Structure determination and Rietveld refinement
The structural determination process was conducted on the basis of previous procedures22,23,25,33 in order to obtain the final crystal structure. The values found via Topas-Academic v.5 were then used in conjunction with the chemical structure (LASSBio-1289 in Fig. 1), in the process of crystal structure determination by means of a simulated annealing algorithm34,35 implemented into the DASH software program.36 Twenty runs (on a total of 2.00 × 107 movements) of the simulated annealing process were globally optimized and the best result was then considered in the Rietveld refinement of the structure.37,38 During the simulated annealing process, the full range of possible values (e.g. 3 degrees of freedom for molecular positions, 4, of which 3 are independent, for orientations as well as 4 flexible torsion angles) was allowed to vary.The background was fitted using a 20-term Chebyshev polynomial. The peak asymmetry was fitted by the simple axial divergence model of Cheary and Coelho.39,40 The peak profiles were modelled by the double-Voigt approach41 with anisotropic peak profiles adjusted using spherical harmonics42 as well as preferred orientation (fourth-order polynomial) of the crystals. Both bond distances and angles and soft planar restraints on rings were restrained to distances and angles mean values obtained from the Mogul Cambridge Structure Databank System.43
Isotropic atomic displacements (Biso) were constrained to be equal for all non-hydrogen atoms. The hydrogen atoms were added considering the orbital geometry using the Mercury software program44 and their Biso values were constrained to be 1.2 times the value of the respective atoms to which they are connected. On the other hand, their fractional coordinates were refined taking into account a macro implemented in Topas-Academic that “rides” (restrains the distances between hydrogen and neighbouring atoms) the H atoms to the ones they are associated to. Initially, its fractional coordinates were inserted as the ones provided by Mercury. Then, taking into account the mean bond distance values found in Mercury (C–H = 0.96 Å, N–H = 0.87 Å and O–H = 0.99 Å), the refinement was performed, thus providing the final atomic coordinates of all atoms.
Fourier transform infrared spectroscopy (FTIR)
Spectral data were recorded in the range from 3000 to 530 cm−1 with a resolution of 4 cm−1, in a Fourier transform spectrometer, model 660-IR, from Varian® (Palo Alto, CA, USA) with an attenuated total reflectance accessory using a diamond crystal. The measurements were conducted at LEMN/UFABC (Laboratory of Electrochemistry and Nanostructured Materials).Nuclear magnetic resonance (NMR)
Both 1H and 13C NMR spectra were determined in deuterated dimethylsulfoxide (DMSO) containing ca. 1% tetramethylsilane as an internal standard, with a Bruker DPX-300 at 300 MHz and 75 MHz, respectively (Bruker BioSpin Co., Billerica, MA, USA). 1H NMR (300 MHz, DMSO-d6) δ 3.53 (s, 3H, N–CH3), 6.05 (s, 2H, OCH2O), 6.87 (d, H–Ar, J = 8.1 Hz), 7.30–7.49 (m, 5H, H–Ar), 7.84 (s, 1H, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH); 13C NMR (75 MHz, DMSO-d6) δ 28.9, 101.4, 107.3, 110.9, 125.2, 125.5, 126.0, 126.6, 128.6, 134.3, 138.2, 146.7, 149.3, 169.8.20
CH); 13C NMR (75 MHz, DMSO-d6) δ 28.9, 101.4, 107.3, 110.9, 125.2, 125.5, 126.0, 126.6, 128.6, 134.3, 138.2, 146.7, 149.3, 169.8.20
Crystal morphology prediction
The prediction and the study of the possible crystal morphologies were performed using a preliminary equilibration protocol, by means of the Discover module included in the Material Studio 7.0 package of Accelrys©, adopting the molecular mechanics approximation and the Compass Force Field (FF).45,46 The cif file containing all the crystallographic information was used as the input for an energy minimization, performed by means of the Smart Minimizer method. The morphology protocol itself is based on the so called GM method. For this purpose, the calculations were carried out in very fine details allowing a minimum interplanar distance (dhkl) of 0.800 Å without setting any limit neither to the values of the three Miller indices nor to the overall number of growing faces.Hartman47 and Perdock,48,49 in order to account for the energy involved in the process related to the definition of a list of potential grown faces and their ability and tendency to grow, introduced the periodic bond chain (PBC) theory, giving rise to the first methodological attempt to rationalize the influence of the energy related to the interactions between growth units. It was therefore possible to give rise to the classification of crystal faces on the basis of the number of PBC belonging to each given adjacent hkl layer – a slice – while the crystallization energy is considered a constant for a given crystal. According to the literature50,51 there are two main contributions to the crystallization energy (Ecr): the energy of each slice (Eslice), i.e. the energy resulting from the lateral interaction of each formula unit within a slice; the attachment energy (Eatt) related to the energy released as a consequence of the vertical interaction of the formula unit with an underlying slice.52 This is summarized as
| Ecr = Eslice + Eatt | (1) |
It was demonstrated48 that
| Gr ∝ Eatt | (2) |
The search for possible solvent accessible voids was performed using the VOID algorithm53,54 setting a grid of 0.20 Å and a probe radius of 1.20 Å.
The Hirshfeld surface was generated by means of the CrystalExplorer software (Build 3.1.1448).55 In the generated map, de is the distance from the Hirshfeld surface to the nearest nucleus outside the surface while di is the corresponding distance to the nearest nucleus inside the surface. We mapped dnorm which is defined as:
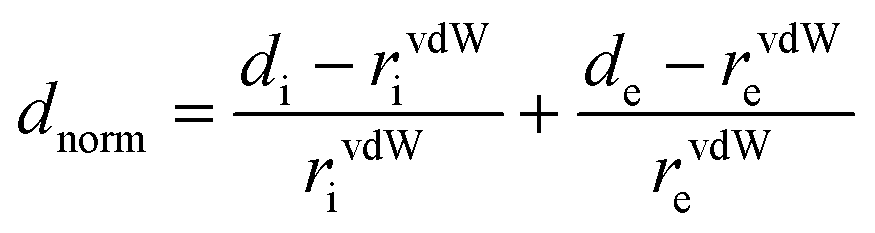 | (3) |
Results and discussion
The final refined values for the unit-cell parameters after the Rietveld fit were: a = 14.5118(3) Å, b = 12.1374(2) Å, c = 7.5498(1) Å, β = 91.113(1)°, V = 1329.53(4) Å3, Z = 4, Z′ = 1 and ρcalc = 1.44042(4) g cm−3. The goodness-of-fit indicator and R-factors were: χ2 = 1.274, RBragg = 1.697%, Rwp = 4.842%, Rexp = 3.802% and Rp = 3.615%. Fig. 2 displays the final Rietveld refinement. | ||
| Fig. 2 Plot from the final Rietveld refinement. Black crosses represent observed data, the red line indicates the calculated pattern and the blue line at the bottom represents the difference between the observed and calculated patterns. Green vertical bars indicate Bragg reflections. After 30° (2θ) the pattern was magnified (5×) for better visualization. | ||
In order to check the consistency of the obtained results for space group choice, unit cell parameters, bond distances, angles and torsions, the PLATON software program was used.56 The ADDSYM routine did not detect any obvious extra crystallographic symmetry. CCDC ID: 1014103 contains the supplementary crystallographic data for this paper.
Crystal data as well as details of the structure determination are shown in Table 1. Structural atomic parameters obtained after the Rietveld refinement are shown in Table 2. Bond distances and bond angles are shown as ESI.†
| Chemical formula | C14H12N2O3S |
| Formula weight (g mol−1) | 288.33 |
| Crystal system | Monoclinic |
| Space group | P21/c (no. 14) |
| a, b, c (Å) | 14.5118(3), 12.1374(2), 7.5498(1) |
| β (°) | 91.113(1) |
| Volume (Å3) | 1329.53(4) |
| Z, Z′ | 4, 1 |
| ρ calc (g cm−3) | 1.44042(4) |
| T (K) | 298 |
| Data collection | |
| Diffractometer | STADI P |
| Monochromator | Ge(111) |
| Wavelength (Å) | 1.54056 |
| 2θ range (°) | 2–60 |
| Step size (°) | 1.05 |
| Time per step (s) | 60 |
| Refinement | |
| Number of data points | 3780 |
| Number of contributing reflections | 368 |
| Number of restraints | 53 |
| Number of refined parameters | 101 |
| R p (%) | 3.615 |
| R exp (%) | 3.802 |
| R wp (%) | 4.842 |
| R Bragg (%) | 1.697 |
| χ 2 | 1.274 |
| LASSBio-1289 υ (cm−1) | Assignment |
|---|---|
| 750 | γ C–H |
| 876 | δ CH out of plane |
| 933 | Dioxolane |
| 1097 | γ C–N and δ C–H in plane |
| 1132 | Dioxolane |
| 1471 | γ C–C |
| 1595 | γ C–C |
| 1639 |
γ CO |
| 1650 |
δ NH in plane and γ CO |
| 2888 | Dioxolane |
The crystal structure of LASSBio-1289 compound consists of four formula units per unit cell (Z = 4), accommodating one molecule in the asymmetric unit (Z′ = 1), which is presented in Fig. 3.
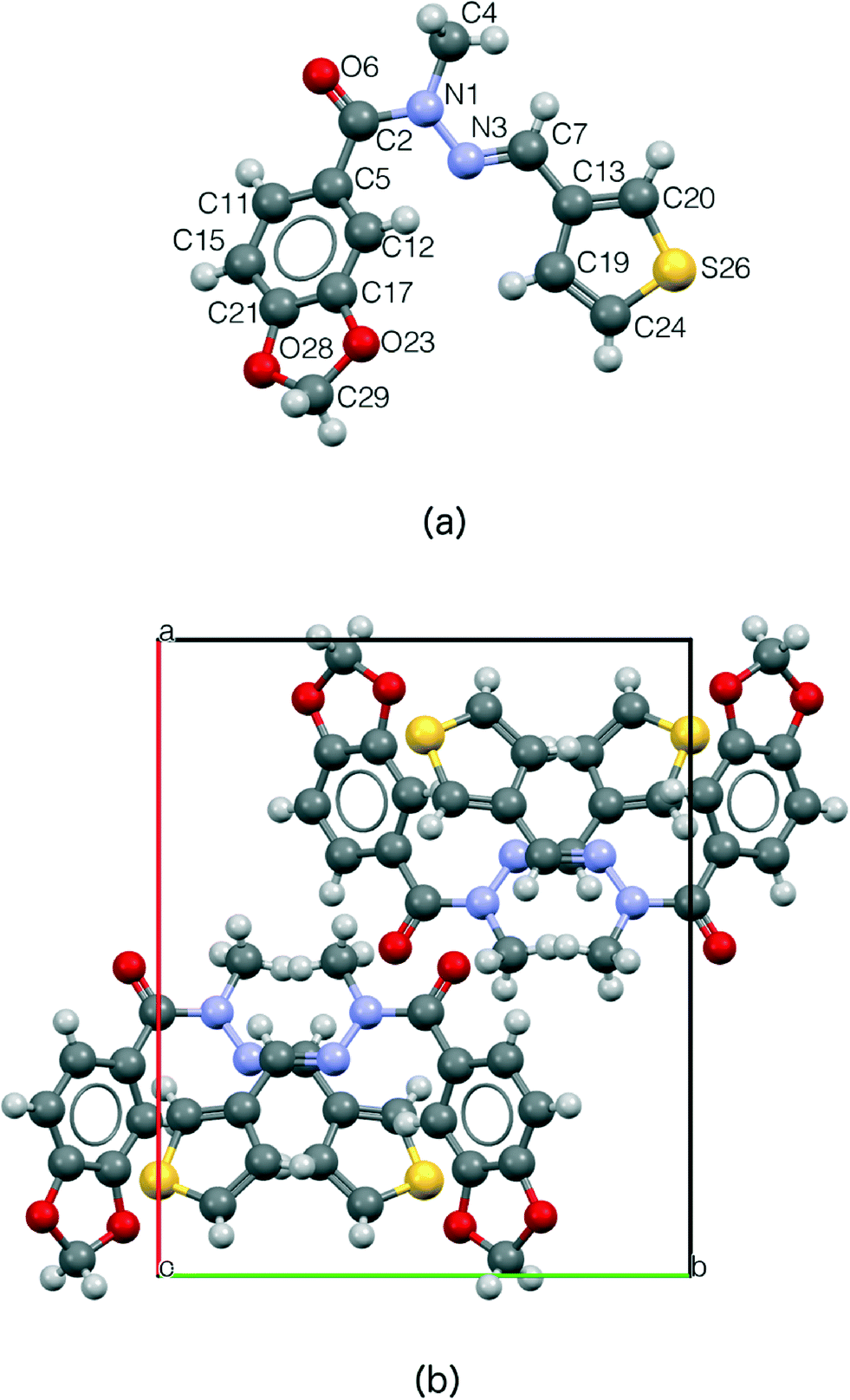 | ||
| Fig. 3 (a) Molecular structure of LASSBio-1289 compound indicating all non-hydrogen atoms as spheres. Light grey spheres represent the hydrogen atoms (not labelled). (b) Unit cell of LASSBio-1289, along the c-axis, with Z = 4. | ||
It is known that N-acylhydrazones may exist as syn/anti amide conformers and Z/E geometrical isomers about the CN double bond.57 The structure determination of LASSBio-1289, by means of X-ray powder diffraction, allowed us to infer the relative configuration E of the imine double bond. This compound presents the amide methyl group syn-periplanar to the carbonyl oxygen, thus forming a folded structure (Fig. 3a) in a similar manner as described in the literature.16,17,58 The adopted syn-periplanar conformation is probably due to a steric effect introduced by the methyl group in relation to the hydrogen present in position 2 of the 1,3-benzodioxolyl ring (Fig. 3).
The methylation effect led to the loss of a hydrogen bond donor. Thus, no significant H-bond was observed. As a consequence, the present structure is an ideal candidate for a crystal morphology prediction.59 Furthermore, we observed some possible dipole–dipole-like interactions (short C–H⋯O contacts). These interactions are involving the amide function and the imino hydrogen of the NAH scaffold (with a distance of 2.454 Å). In addition, we also observed some possible auxiliary T-shape interactions between the hydrogen of thiophenyl ring and 1,3-benzodioxolyl ring (2.746 Å) and between the hydrogen present in position 2 of the 1,3-benzodioxolyl ring and the thiophenyl ring (2.946 Å). Indeed, these non-polar interactions seem to contribute to the organization of the space arrangement in the unit cell, especially as a consequence of the absence of strong interactions, as shown in Fig. 4. In Fig. 4a, there is a quantitative description of the above-mentioned contacts. A Hirshfeld surface map is reported in Fig. 4b in order to enable their fast visual evaluation.
 | ||
| Fig. 4 (a) Some non-covalent interactions of LASSBio-1289 (green-dashed lines) contributing for the organization of the space arrangement within the unit cell. Measured distances (Å) are highlighted in green-dashed lines. (b) Hirshfeld surface, generated as described in the Experimental section. Atoms involved in intermolecular contacts closer than the sum of their van der Waals radii are highlighted in red; longer contacts are indicate in blue and contacts around the sum of van der Waals radii are displayed in white. | ||
Fourier transform infrared spectroscopy
Fig. 5 displays the FTIR spectrum of LASSBio-1289. Some characteristic bands are indicated in Table 2. | ||
| Fig. 5 FTIR spectrum of LASSBio-1289 sample obtained with a resolution of 4 cm−1 in the region from 3000 to 530 cm−1. | ||
The dioxolane displays two peaks of medium intensity at 939 cm−1 and 1117 cm−1 and another one, of lower intensity, at 2888 cm−1.60 The absorption bands of CO from amides were found at wavelengths greater than the observed ones for the carbonyl due to the resonance effect. The primary amide shows an intense band at 1650 cm−1. The absorption of the CO group occurs between 1680 cm−1 and 1630 cm−1.60 The most important bands, thus providing more information about the structure of the aromatic compounds, are found in the region of low frequencies, where one can observe a vibration at 751 cm−1. This intense band arises from the angular deformation of the C–H bonds out of the plane of the ring. The angular deformation band in plane appears at 1045 cm−1. Vibrations are observed at 1600–1585 cm−1 and 1500–1400 cm−1, involving the axial deformation of the C–C bonds of the ring.60
In primary aliphatic amine, the absorption band of the unconjugated C–N bond appears at 1250 cm−1. This band has low-medium intensity, originating from the axial deformation of the C–N group and is coupled to the axial deformation of the adjacent connections of the molecule. The thiophene group, belonging to the class of the heteroaromatics, has an axial deformation band of C–H at 3021 cm−1.60
Nuclear magnetic resonance
Our structural analysis was also performed in solution, in order to evaluate whether the main structural features already highlighted in the solid state were kept almost unaltered.The absence of duplicated signals in 1H and 13C-NMR data (see ESI†) confirmed that the title compound was obtained as a single diastereoisomer and conformational state in solution. The relative configuration of the imine unit of LASSBio-1289 was defined as (E) and the amide bond of NAH moiety seems to present the syn-periplanar conformation, as anticipated in our previous works16,61 with N-methyl-N-acylhydrazone derivatives and corroborated by the X-ray diffraction studies described herein.
There is evidence, therefore, of an interesting structural agreement between solid and liquid phases.
Crystal morphology and scanning electron microscopy
Fig. 6 shows the acquired scanning electron microscopy (JSM-6010LA, from JEOL® (Tokyo, Japan), located at the Multiuser Experimental Centre of Federal University of ABC (CEM/UFABC)) image of some LASSBio-1289 crystals, exhibiting a needle-like shape and relatively smooth surfaces. In general, the crystals have thicknesses up to 10 μm and longer lengths. | ||
| Fig. 6 Scanning electron microscopy image of some LASSBio-1289 crystals exhibiting needle-like shapes and relatively smooth surfaces. | ||
A preliminary crystal morphology prediction was performed using the BFDH algorithm,62–64 which takes into account geometrical considerations only and reported in the ESI.† In Fig. 7, the morphology calculated as described in the Experimental section is reported, while Table 3 lists the characteristics of the Morphologically Important (MI) faces.
 | ||
| Fig. 7 Crystal morphology prediction of LASSBio-1289. Miller indices of the MI faces are reported. | ||
| hkl | Multiplicity | d hkl (Å) | E att (total) (kcal mol−1) | Total facet area (%) |
|---|---|---|---|---|
| (100) | 2 | 14.5091 | −23.0337 | 46.84 |
| (110) | 4 | 9.3095 | −44.6634 | 27.26 |
| (011) | 4 | 6.4099 | −67.2165 | 8.86 |
| (11−1) | 4 | 5.8993 | −64.7692 | 17.04 |
Interestingly, if we would have limited the morphologically prediction to pure geometrical considerations – such as in the BFDH approach – we would have only assumed that the growth rate of the crystal face is inversely proportional to the interplanar spacing dhkl. As a consequence, the MI faces of the crystals would have been those having the largest dhkl values.62,63 However, this approach does not consider the energetics of the systems. On the other hand, by using the GM approach, we take into account the Eatt, thus simulating the crystal habit as specified in the Experimental section. It is on this basis that we can interpret the apparently inconsistent and swapped TFA percentages for (001) and (11−1): the higher (011) Eatt values are in fact counterbalanced by the greater interplanar distance dhkl. As reported in the ESI,† the BFDH method highly underestimated the role of (11−1).
With these premises, at a first glance, there is not a complete agreement between the morphology prediction results and the crystals micrograph shown in Fig. 6. However, the calculations were performed in vacuo while the “real” crystals were grown interacting with solvents. For this reason, we decided to perform a detailed analysis of the potential role of the crystallization solvents. This study will give us key information on one hand about the reasons why some MI faces are overgrown or underestimated by the calculation; on the other hand it will help us making some practical hypotheses about the solubility of these crystals.65,66
For this purpose, in Fig. 8 we analysed the results of the computed morphology, drawing the hkl plane corresponding to the MI crystal faces reported in the habit, simply taking into account which kind of groups are overlooking the considered face.
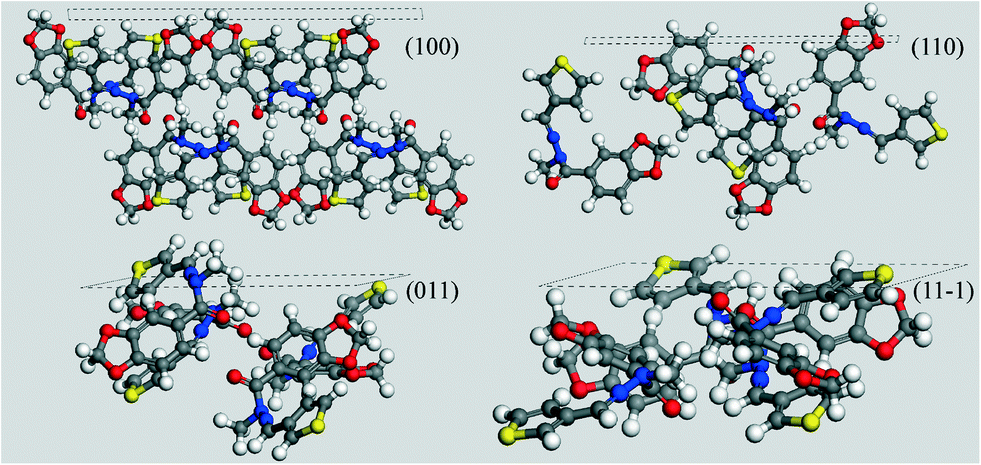 | ||
| Fig. 8 MI faces for LASSBio-1289. The relative Miller indices are shown together with the respective outcropping atoms. | ||
The structure shows a high packing index (69.8%)54 and no residual solvent accessible voids. As a consequence, we can assume LASSBio-1289 is unable to host solvents without modifying its crystal structure and the attachments of solvent molecules must take place directly from the outer part of the considered faces. This is true whatever crystallization mechanism is considered, neither the classical nucleation theory nor the two-step mechanism.67,68
With the exception of the (100) face, all the other reported MI faces evidence polar groups or moieties. As the final flash crystallization of the compound was performed by means of cold water, the use of this extremely polar solvent inhibited the growth of all the MI faces but the (100). Since it shows mainly apolar groups and atoms to the crystal surface, its growth was left unaltered – or it was possibly enhanced – as compared to the other MI faces. As a consequence, we can easily assume that thin needles with a predominant elongation along this overgrown face can be expected, in a very good agreement with the microcrystals reported in Fig. 6.
Conclusions
From the crystal structure determination we could infer some possible dipole–dipole-like and auxiliary T-shape interactions, which contribute to the organization of the LASSBio-1289 molecules within the unit cell. The amide methyl group is in a syn-periplanar orientation in relation to the carbonyl oxygen, thus forming a folded structure. The morphology prediction evidenced the presence of differences between the theoretical model and the experimental one. They have been interpreted on the basis of the role of the crystallization solvent. Interestingly, the NMR measurements confirmed in solution a layout compatible with the one depicted in the solid state.Acknowledgements
We thank the financial support provided by the São Paulo State Foundation (FAPESP, grant no. 2008/10537-3), the National Council for Scientific and Technological Development (CNPq, grants no. 305186/2012-4 and 141308/2012-5), CAPES (grant no. 88881.062195/2014-01), INCT-INOFAR (BR, # 573.564/2008-6), FAPERJ and UFABC.References
- J. He and P. K. Whelton, Med. Clin. North Am., 1997, 81, 1077–1097 CrossRef CAS.
- P. K. Whelton, Lancet, 1994, 344, 101–106 CrossRef CAS.
- M. Ezzati, A. D. Lopez, A. Rodgers, S. V. Hoorn, C. J. L. Murray and C. R. A. C. Group, Lancet, 2002, 360, 1347–1360 CrossRef.
- A. Chockalingam, N. R. Campbell and J. G. Fodor, Can. J. Cardiol., 2006, 22, 553–555 CrossRef.
- P. M. Kearney, M. Whelton, K. Reynolds, P. K. Whelton and J. He, J. Hypertens., 2004, 22, 11–19 CrossRef CAS PubMed.
- B. V. Mittal and A. K. Singh, Am. J. Kidney Dis., 2010, 55, 590–598 CrossRef PubMed.
- K. B. Tibazarwa and A. A. Damasceno, Can. J. Cardiol., 2014, 30, 527–533 CrossRef PubMed.
- J. W. Levenson, P. J. Skerrett and J. M. Gaziano, Prev. Cardiol., 2002, 5, 188–199 CrossRef PubMed.
- P. M. Kearney, M. Whelton, K. Reynolds, P. Muntner, P. K. Whelton and J. He, Lancet, 2005, 365, 217–223 CrossRef.
- L. E. Fields, V. L. Burt, J. A. Cutler, J. Hughes, E. J. Roccella and P. Sorlie, Hypertension, 2004, 44, 398–404 CrossRef CAS PubMed.
- L. Paulis, U. M. Steckelings and T. Unger, Nat. Rev. Cardiol., 2012, 9, 276–285 CrossRef CAS PubMed.
- H. Gonzalez-Serratos, R. Chang, E. F. R. Pereira, N. G. Castro, Y. Aracava, P. A. Melo, P. C. Lima, C. A. M. Fraga, E. J. Barreiro and E. X. Albuquerque, J. Pharmacol. Exp. Ther., 2001, 299, 558–566 CAS.
- C. L. M. Silva, F. Noël and E. J. Barreiro, Br. J. Pharmacol., 2002, 135, 293–298 CrossRef CAS PubMed.
- A. G. Silva, G. Zapata-Sudo, A. E. Kummerle, C. A. M. Fraga, E. J. Barreiro and R. T. Sudo, Bioorg. Med. Chem. Lett., 2005, 13, 3431–3437 CrossRef CAS PubMed.
- R. T. Sudo, G. Zapata-Sudo and E. J. Barreiro, Br. J. Pharmacol., 2001, 134, 603–613 CrossRef CAS PubMed.
- A. E. Kümmerle, J. M. Raimundo, C. M. Leal, G. S. da Silva, T. L. Balliano, M. A. Pereira, C. A. de Simone, R. T. Sudo, G. Zapata-Sudo, C. A. M. Fraga and E. J. Barreiro, Eur. J. Med. Chem., 2009, 44, 4004–4009 CrossRef PubMed.
- E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed.
- G. Zapata-Sudo, S. L. Pereira, H. J. V. Beiral, A. E. Kümmerle, J. M. Raimundo, F. Antunes, R. T. Sudo, E. J. Barreiro and C. A. Fraga, Am. J. Hypertens., 2010, 23, 135–141 CrossRef CAS PubMed.
- L. M. Lima and E. J. Barreiro, Curr. Med. Chem., 2005, 12, 23–49 CrossRef CAS.
- S. L. Pereira, A. E. Kümmerle, C. A. Fraga, E. J. Barreiro, R. T. Sudo and G. Zapata-Sudo, Fundam. Clin. Pharmacol., 2014, 28, 29–41 CrossRef CAS PubMed.
- S. L. Pereira, A. E. Kümmerle, C. A. Fraga, E. J. Barreiro, N. N. Rocha, E. B. Ferraz, J. H. do Nascimento, R. T. Sudo and G. Zapata-Sudo, Eur. J. Pharmacol., 2013, 702, 316–322 CrossRef CAS PubMed.
- F. F. Ferreira, S. G. Antonio, P. C. P. Rosa and C. O. Paiva-Santos, J. Pharm. Sci., 2010, 99, 1734–1744 Search PubMed.
- F. F. Ferreira, A. C. Trindade, S. G. Antonio and C. O. Paiva-Santos, CrystEngComm, 2011, 13, 5474–5479 RSC.
- A. J. Florence, N. Shankland, K. Shankland, W. I. F. David, E. Pidcock, X. Xu, A. Johnston, A. R. Kennedy, P. J. Cox, J. S. O. Evans, G. Steele, S. D. Cosgrove and C. S. Frampton, J. Appl. Crystallogr., 2005, 38, 249–259 CrossRef CAS.
- F. N. Costa, F. F. Ferreira, T. F. da Silva, E. J. Barreiro, L. M. Lima, D. Braz and R. C. Barroso, Powder Diffr., 2013, 28, S491–S509 CrossRef CAS.
- F. N. Costa, D. Braz, F. F. Ferreira, T. F. da Silva, E. J. Barreiro, L. M. Lima, M. V. Colaço, L. Kuplich and R. C. Barroso, Radiat. Phys. Chem., 2014, 95, 292–295 CrossRef CAS PubMed.
- G. Li Destri, A. Marrazzo, A. Rescifina and F. Punzo, J. Pharm. Sci., 2011, 100, 4896–4906 CrossRef CAS PubMed.
- G. Li Destri, A. Marrazzo, A. Rescifina and F. Punzo, J. Pharm. Sci., 2013, 102, 73–83 CrossRef PubMed.
- A. A. Coelho, J. Evans, I. Evans, A. Kern and S. Parsons, Powder Diffr., 2011, 26, s22–s25 CrossRef CAS.
- A. A. Coelho, J. Appl. Crystallogr., 2003, 36, 86–95 Search PubMed.
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- B. H. Toby, Powder Diffr., 2006, 21, 67–70 CrossRef CAS.
- A. Gomez, S. G. Antonio, G. L. B. de Araujo, F. F. Ferreira and C. O. Paiva-Santos, CrystEngComm, 2012, 14, 2826–2830 RSC.
- E. Aarts and J. Korst, Simulated Annealing and Boltzmann Machines: a Stochastic Approach to Combinatorial Optimization and Neural Computing, ed. E. Aarts and J. Korst, John Wiley & Sons, Chichester, UK, 2nd edn, 1991 Search PubMed.
- P. J. M. van Laarhoven and E. H. L. Aarts, Simulated Annealing: Theory and Applications, ed. P. J. M. van Laarhoven and E. H. L. Aarts, Kluwer Academic Publishers, Dordrecht, Holland, 4th edn, 1992 Search PubMed.
- W. I. F. David, K. Shankland, J. V. D. Streek, E. Pidcock, W. D. S. Motherwell and J. C. Cole, J. Appl. Crystallogr., 2006, 39, 910–915 CrossRef.
- H. M. Rietveld, Acta Crystallogr., 1967, 22, 151–152 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- R. W. Cheary and A. A. Coelho, J. Appl. Crystallogr., 1998, 31, 851–861 CrossRef CAS.
- R. W. Cheary and A. A. Coelho, J. Appl. Crystallogr., 1998, 31, 862–868 CrossRef CAS.
- D. Balzar, J. Res. Natl. Inst. Stand. Technol., 1993, 98, 321–353 CrossRef CAS.
- M. Järvinen, J. Appl. Crystallogr., 1993, 26, 525–531 CrossRef.
- I. J. Bruno, J. C. Cole, M. Kessler, J. Luo, W. D. S. Motherwell, L. H. Purkis, B. R. Smith, R. Taylor, R. I. Cooper, S. E. Harris and A. G. Orpen, J. Chem. Inf. Comput. Sci., 2004, 44, 2133–2144 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef.
- S. Huai, S. J. Mumby, J. R. Maple and A. T. Hagler, J. Am. Chem. Soc., 1994, 116, 2978–2987 CrossRef.
- P. Hartman, J. Cryst. Growth, 1980, 49, 157–165 CrossRef CAS.
- P. Hartman and W. G. Perdock, Acta Crystallogr., 1955, 8, 49–52 CrossRef.
- P. Hartman and P. Bennema, J. Cryst. Growth, 1980, 49, 145–156 CrossRef.
- R. Docherty, G. Clydesdale, K. J. Roberts and P. Bennema, J. Phys. D: Appl. Phys., 1991, 24, 89–99 CrossRef CAS.
- F. Punzo, Cryst. Growth Des., 2011, 11, 3512–3521 CAS.
- Z. Berkovitch-Yellin, J. Am. Chem. Soc., 1985, 107, 8239–8253 CrossRef CAS.
- P. Van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- A. I. Kitajgorodskij, Molecular Crystals and Molecules, Academic Press, New-York, 1st edn, 1973 Search PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- G. Palla, G. Predieri, P. Domiano, C. Vignali and W. Turner, Tetrahedron, 1986, 42, 3649–3654 CrossRef CAS.
- H. Kagechika, T. Himi, E. Kawachi and K. Shudo, J. Med. Chem., 1989, 32, 2292–2296 CrossRef CAS.
- F. Punzo, J. Mol. Struct., 2013, 1032, 147–154 CrossRef CAS PubMed.
- R. M. Silverstein, F. X. Webster and D. J. DKiemle, Spectrometric identification of organic compounds, John Wiley & Sons, Inc., Westford, MA, USA, 7th edn, 2005 Search PubMed.
- A. B. Lopes, E. Miguez, A. E. Kümmerle, V. M. Rumjanek, C. A. M. Fraga and E. J. Barreiro, Molecules, 2013, 18, 11683–11704 CrossRef PubMed.
- M. A. Bravais, Études Cristallographiques, Gauthier Villars, Paris, France, 1866 Search PubMed.
- G. Friedel, Bull. Soc. Fr. Mineral., 1907, 30, 326–455 Search PubMed.
- J. D. H. Donnay and D. Harker, Am. Mineral., 1937, 22, 446–467 Search PubMed.
- A. R. Lazo Fraga, F. F. Ferreira, G. M. Lombardo and F. Punzo, J. Mol. Struct., 2013, 1047, 1–8 CrossRef PubMed.
- G. M. Lombardo, A. Rescifina, U. Chiacchio, A. Bacchi and F. Punzo, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 172–180 CAS.
- P. G. Vekilov, Cryst. Growth Des., 2007, 7, 2796–2810 CAS.
- P. G. Vekilov, Cryst. Growth Des., 2010, 10, 5007–5019 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectrum, X-ray crystallographic data in CIF format, tables for final atomic structural parameters together with some selected bond lengths and bond angles as well as the results of the BFDH morphology prediction. CCDC 1014103. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce02011a |
| This journal is © The Royal Society of Chemistry 2015 |