
Shaping calcite crystals by customized self-assembling pseudopeptide foldamers†
Claudia
Tomasini
*,
Nicola
Castellucci
,
Valentina C.
Caputo
,
Lorenzo
Milli
,
Giulia
Battistelli
,
Simona
Fermani
and
Giuseppe
Falini
*
Dipartimento di Chimica “Giacomo Ciamician”, Alma Mater Studiorum – Università di Bologna, via Selmi 2, I-40126 Bologna, Italy. E-mail: giuseppe.falini@unibo.it; claudia.tomasini@unibo.it
First published on 27th October 2014
Abstract
Two pseudopeptide foldamers with similar backbones, one containing a D-4-carboxy-5-methyl-oxazolidin-2-one moiety (1) and the other a D-proline moiety (2), self-assembled in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water–ethanol mixture. Molecule 1 formed fibres that generated a highly viscous sol or a gel by increasing its concentration; molecule 2 assembled in nanoparticles that aggregated in bigger particles by increasing its concentration. This behaviour was conserved in the presence of 10 mM CaCl2. Both foldamers, which exposed carboxylate groups, were able to modify the shape of single crystals of calcite. The presence of molecule 1 favoured the formation of rhombohedral calcite showing additional crystalline faces, while molecule 2 induced the formation of cavities and curvatures. Thus, pseudopeptide foldamers diversely act as crystal growth modifiers according to minor structural changes that mutate their self-assembly. This result is of general interest for the design of new molecules affecting the crystallization process and has implications in understanding how biological molecules control the growth of mineral phases.
:1 water–ethanol mixture. Molecule 1 formed fibres that generated a highly viscous sol or a gel by increasing its concentration; molecule 2 assembled in nanoparticles that aggregated in bigger particles by increasing its concentration. This behaviour was conserved in the presence of 10 mM CaCl2. Both foldamers, which exposed carboxylate groups, were able to modify the shape of single crystals of calcite. The presence of molecule 1 favoured the formation of rhombohedral calcite showing additional crystalline faces, while molecule 2 induced the formation of cavities and curvatures. Thus, pseudopeptide foldamers diversely act as crystal growth modifiers according to minor structural changes that mutate their self-assembly. This result is of general interest for the design of new molecules affecting the crystallization process and has implications in understanding how biological molecules control the growth of mineral phases.
Introduction
The synthesis of complex materials with excellent properties for modern technologies requires innovative approaches. Biomineralization takes advantage of elaborate biological processes, which produce biominerals with complex and fascinating morphologies as well as superior mechanical properties.1,2 The customized production of inorganic materials of specific morphology and size is a key goal in the fields of advanced materials, electronics, medicine, catalysis, ceramics, cosmetics and pigments.3 Calcium carbonate particles are an important class of materials because their unique structural, optical and surface properties can lead to a wide range of application.4 During the past decades numerous organic templates or modifiers with complex functional patterns to control the nucleation, growth, and alignment of inorganic crystals have been widely used for the biologically inspired synthesis of inorganic materials with complex form.5 This includes folded peptides,6 biopolymers,7 self-assembled monolayers,8 other supramolecular assemblies,9 and small molecules.10Since many macromolecules involved in biomineralization are rich in aspartic and glutamic residues, a common strategy in the synthesis of crystal morphology modifiers involves the design of molecules with ordered sequences of negatively charged functional groups (e.g. carboxylates and phosphates).11
Pseudopeptide foldamers are synthetic molecules that can organize into well-defined secondary structures (i.e. helices, turns and sheets). Despite their small size, they closely mimic biomacromolecules.12 Their repetitive secondary structure is imparted by conformational restrictions of the monomeric unit.13 Foldamers designed to expose charged functional groups in specific stable folding can interact with ordered array of ions of crystalline planes and act as effective crystal growth modifiers.14
Peptide-based polyisocyanides were used as organic templates for the crystallization of calcium carbonate. These molecules adopted a helical conformation due to restricted rotation of the polymer backbone and H bonding interactions between dipeptide side chains.15 The polydispersity and the molecular weight of the peptides were shown to have no effect on the stabilization of {hk.0} faces of calcite that was nucleated on the (01.1) plane. Ueyama et al. showed that in the presence of aromatic polyamide foldamers with ordered sequences of carboxylate groups, the growth of the {10.4} faces in calcite was drastically reduced. Moreover, the aromatic polyamide molecules entrapped in the calcite crystals were detected by a NMR technique.16 A synthetic tricarboxylic acid foldamer derivative projecting carboxylates in an ordered fashion was used to influence the growth of calcium carbonate. A specific interaction between the growing calcite crystals and this foldamer resulted in the formation of new {10.l} faces and a unique morphology.17
In the past few years we have extensively studied the conformational behaviour of foldamers containing the 4-carboxy-5-methyl-oxazolidin-2-one (Oxd) moiety.18 Acylation of the Oxd units yields imides that behave like rigid spacers, and the relative conformation of the two carbonyls generates a trans conformation with respect to the adjacent peptide bond.19 We have also demonstrated that the protected pseudopeptide foldamer Boc-L-Phe-D-Oxd-OBn forms a fiber-like material by self-organization as it spontaneously forms infinite linear chains where the parallel dipeptide units are connected only by single hydrogen bonds.20
To understand the importance of the L-Phe-D-Oxd scaffold to build new architectures, we have decided to explore how it behaves when it is included in more complex structures, which can specifically modify the growth of calcite crystals according to their capability of self-assembly.
We selected CH2(C3H6CO-L-Phe-D-Oxd-OH)21 that we have recently prepared. It is an efficient and biocompatible low molecular weight gelator (LMWG) that has been used as a “Trojan horse” for drug delivery.21 It is known that three factors can favour the formation of a gel: (i) the presence of hydrogen bonding and π–π stacking interactions, which are the principle interactions involved in gel aggregation; (ii) the tendency of the molecule head to organize into a network that influences the probability of gel formation; and (iii) the presence of a medium-sized aliphatic chain (4–8 methylene units) connected with a polar head.22
Simultaneously we have prepared CH2(C3H6CO-L-Phe-D-Pro-OH)22 that has the same structure, but the D-Oxd moiety has been replaced with a D-Pro (D-Pro = D-proline) moiety to check if the presence of the Oxd moiety is essential for the existence of the properties of the supramolecular material (Fig. 1).
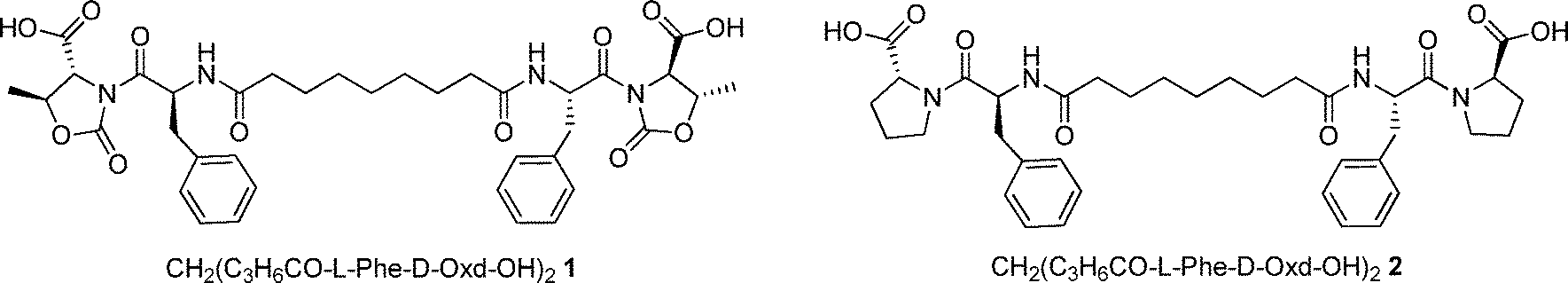 | ||
| Fig. 1 Chemical structures of molecules 1 and 2. | ||
Results and discussion
Synthesis of 1 and 2 molecules and their supramolecular assembly
Molecules 1 and 2 are long-chain derivatives and belong to the family of bolaamphiphiles.23 Synthetic bolaamphiphiles try to reproduce the unusual architecture of monolayered membranes found in archaebacteria but commonly do not use the same building blocks, which are difficult to synthesize. The synthesis of these molecules has been previously described and may be easily obtained on a multigram scale.24The study on the self-assembly capability of molecules 1 and 2 showed that while molecule 2 does not form a gel at any concentration, molecule 1 forms a gel at 10 mM concentration and a highly viscous sol at 5 mM concentration (Fig. 2A, B). A detailed study on the morphology of pure hydrogel 1 has been previously reported.25
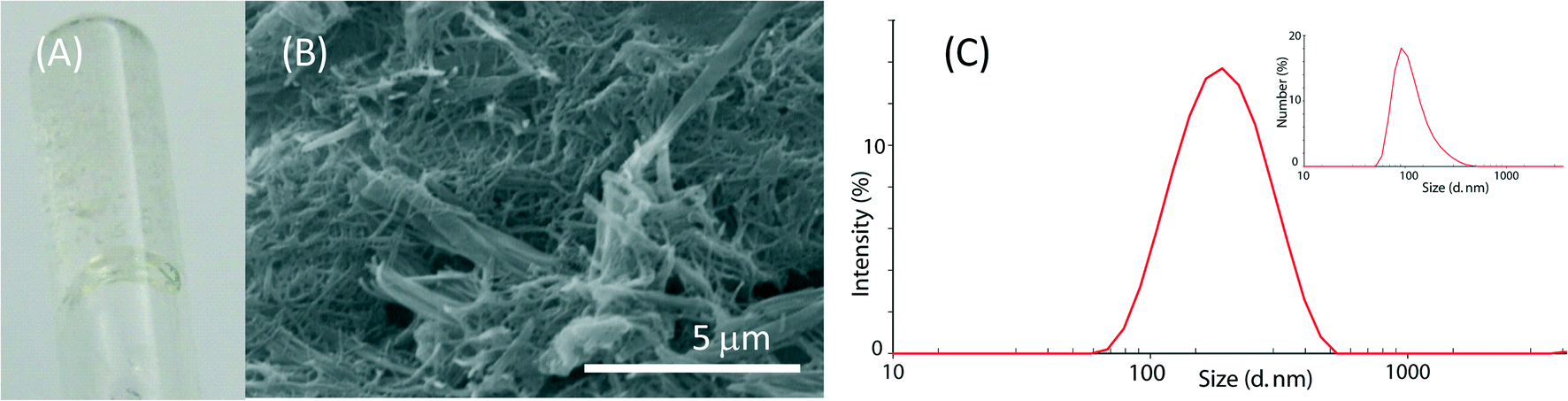 | ||
| Fig. 2 (A) Photograph of hydrogel obtained from a 10 mM solution of molecule 1 in a 10 mM CaCl2 solution in a 9:1 (v/v) H2O–EtOH mixture. (B) Scanning electron microscopy picture of the corresponding xerogel. (C) Size distribution by intensity (by number in the inset) of the particles from a 1 mM solution of molecule 2 in a 10 mM CaCl2 solution in a 9:1 (v/v) H2O–EtOH mixture. | ||
The assembly of molecule 2 was investigated by dynamic light scattering. This molecule assembled in structures with a diameter ranging around 100 nm in a 1 mM solution (Fig. 2C). By increasing the molecule 2 concentration, the solutions became opalescent. Upon 0.45 μm filtration, clear solutions were obtained and only particles with a size around 100 nm were observed in the 1 mM and the 5 mM solutions (Fig. S1†), while in the 10 mM solution, mainly small particles of about 1 nm were observed. This information suggests that at a concentration of 10 mM, the 100 nm particles mainly aggregate to form bigger structures that are removed by 0.45 μm filtration, thus leaving in solution almost only single molecules with a size of about 1 nm. Besides the characterization on molecule 2 aggregation as a function of its concentration, molecule 2 solutions were used for the CaCO3 precipitation experiments without applying any filtration procedure.
CaCO3 precipitation experiments
The presence of EtOH causes a reduction in the solubility of CaCO3. This implies that nucleation and growth processes occur at lower values of supersaturation than in pure water. The presence of EtOH also causes a reduction in the hydration sphere of the ions. In this process, the solvation enthalpy does not change significantly, while it is significantly reduced in the perturbation of the ion in the structure of the solvent. Thus, the reduction in the hydration sphere in the presence of EtOH is mainly due to an entropic factor. This facilitates the crystal nucleation and growth process as the ion incorporation into the crystalline structure requires de-solvation.28
In agreement with the above considerations, from both 10 mM CaCl2 and 100 mM CaCl2 in a 9/1 (v/v) H2O–EtOH mixture, the precipitation of calcite, the most stable CaCO3 polymorph, was exclusively observed (Fig. S2†). The induced nucleation of CaCO3 causes a decrease in the average size of the crystals and an increase in density of precipitation because the crystal nuclei number increased. The Ca2+ mass balance determined the stop of the growth and the average size of the crystals.
Calcite precipitated forming perfect rhombohedra that showed only {10.4} faces, and the size of the crystals was from 10 to 100 μm and from 5 to 50 μm of the 10 mM CaCl2 (low supersaturation) and the 100 mM CaCl2 (high supersaturation) solutions, respectively (Fig. 3).
 | ||
| Fig. 3 Optical and scanning electron microscopy pictures of calcite crystals precipitated from the 9/1 (v/v) H2O–EtOH mixture from a 10 mM (A, B) or 100 mM (C, D) CaCl2 solution. These pictures are representative of the entire population of crystals. | ||
:1 H2O–EtOH mixture. Several concentrations of both molecules have been analyzed: 1 mM, 5 mM or 10 mM. For molecule 2, the study was also carried out in a 100 mM CaCl2 solution, as molecule 1 is not soluble in the 100 mM CaCl2 solution. The X-ray diffraction analysis of all the samples showed diffraction patterns in which only peaks associable to calcite were observed (Fig. S3 and S4†).
 | ||
| Fig. 4 Optical (A, D) and scanning electron microscopy (B, C, E, F) pictures of calcite crystals precipitated from the 9/1 (v/v) H2O–EtOH mixture in the presence of 1 mM molecule 1 (A–C) or molecule 2 (D–F). These pictures are representative of the entire population of crystals. | ||
Molecule 1 forms a highly viscous sol at 5 mM with a Ca2+/molecule 1 molar ratio of 2. Here, the molecule 1 effect on the nucleation and growth of CaCO3 is driven by the presence of supramolecular assemblies in solution. Optical microscopy images (Fig. 5A) of the precipitated particles under these conditions show that they are single crystals with an average size of about 30 μm, which is smaller with respect to the crystals precipitated in the absence of molecule 1. This effect may be due to an inhibition of the growth process, which involves the adsorption of the assembled molecule 1 on the crystals. The crystals show a non-homogeneous diffusion of visible light, with dark areas in the central region of the faces, where the adsorption is supposed to occur. The SEM image in Fig. 5B shows an aggregate of molecule 1 forming needle-like units located on the CaCO3 crystal face. In the SEM image in Fig. 5C, an aggregate of molecule 1 is partially entrapped in the CaCO3 crystal. These aggregates can act as a site for the heterogeneous nucleation of crystals of CaCO3 and becomes completely entrapped in the crystals (dark areas on the crystals in the optical microscope images). It is also noted that the rhombohedral morphology of the crystals is altered. In the crystals {18.0} faces were shown in addition to the {10.4} faces. The {18.0} faces should appear upon interaction of molecule 1 with these crystalline planes.
 | ||
| Fig. 5 Optical (A, D) and scanning electron microscopy (B, C, E, F) pictures of calcite crystals precipitated from the 9/1 (v/v) H2O–EtOH mixture in the presence of 5 mM molecule 1 (A–C) or molecule 2 (D–F). These pictures are representative of the entire population of crystals. | ||
At a concentration of 10 mM, molecule 1 forms gels. Under these conditions, with a Ca2+/molecule 1 molar ratio of 1, the effect on the nucleation and growth of CaCO3 is due to the presence of fibrous structures from molecule 1 (Fig. 2B). Here, the crystal growth occurs in the confined space generated by the three-dimensional fibrous network making the gel. In this medium, the ion diffusion is slower than that in solution and convection is absent. This determines a slower process of crystal growth. The optical microscope images (Fig. 6A) of the precipitated particles show that they are single crystals with an average size of about 50 μm, which is bigger with respect to the crystals precipitated in the absence of molecule 1. This effect may be due to an inhibition of the nucleation process. The crystals do not scatter light, suggesting that the gel fibres are entrapped in the crystalline structure. A similar observation has been reported for the growth of calcite in agarose gel.29 The SEM image (Fig. 6B) shows the co-precipitation of CaCO3 crystals of varying shapes. These are probably due to structural reorganization of the gel regions that occurred during the process of crystallization. The SEM image at higher magnification (Fig. 6C) illustrates how the CaCO3 crystals have almost completely lost the rhombohedral morphology. In the crystals, the {10.4} faces are little exposed and the {kh.0} faces appear more extended, indicating an interaction of molecule 1 with these families of crystallographic planes. The FTIR spectra (Fig. S5†) are in agreement with the above observations; in fact absorption bands due to molecule 1 and calcite were observed. The relative intensity of the absorption bands due to molecule 1 increases with its concentration in solution. Unfortunately, the co-presence of calcite crystals and of an additional phase containing molecules 1 does not allow quantification of molecule 1 entrapped in the crystals.
 | ||
| Fig. 6 Optical (A, D) and scanning electron microscopy (B, C, E, F) pictures of calcite crystals precipitated from the 9/1 (v/v) H2O–EtOH mixture in the presence of 10 mM molecule 1 (A–C) or molecule 2 (D–F). These pictures are representative of the entire population of crystals. | ||
Fig. 4D–E illustrate the optical and electron microscope images of the precipitate obtained in the presence of 1 mM molecule 2 (Ca2+/molecule 2 molar ratio = 10). The optical microscope images (Fig. 4E) of the precipitated particles show that they are single crystals (Fig. S5†) with an average size of about 40 μm, which are similar to the crystals precipitated in the absence of molecule 2. The SEM images (Fig. 4E, F) show that the rhombohedral crystal morphology was not significantly altered. However, the crystals showed on the {10.4} faces some zones of interaction with the molecule characterized by considerable roughness. This interaction appears as a surface phenomenon on the {10.4} faces suggesting that molecule 2 affected the crystal growth process without specifically interacting with some crystalline planes. This nonspecific interaction generated in some crystals the appearance of {kh.0} faces together with the rhombohedral {10.4} faces.
Fig. 5D–E show the optical and electron microscope images of the precipitate obtained in the presence of 5 mM molecule 2 (Ca2+/molecule 2 molar ratio = 2). The optical microscope images (Fig. 5D) show single-crystal particles that are not aggregated and with an average size of about 40 μm, the same as those precipitated in the absence of molecule 2. SEM images (Fig. 5D, E) showed that in the rhombohedral crystals, zones characterized by considerable roughness were observed on the {10.4} faces. Moreover, in some crystals, cavities with sub-micrometer dimensions (Fig. 5E) were also observed on the same faces. These cavities appeared as dark regions under the optical microscope. This crystallization condition also caused the formation of shapeless aggregates (Fig. 5E) probably due to the precipitation of molecule 2.
The optical and electron microscope images of the precipitate obtained in the presence of 10 mM molecule 2 (Ca2+/molecule 2 molar ratio = 1) are illustrated in Fig. 6D–E. The optical microscopy image (Fig. 6D) shows that crystals are aggregated and each crystal has an average size of about 20 μm. SEM images (Fig. 6E, F) show that on the {10.4} faces, cavities along the direction parallel to the rhombohedral axis were present. These cavities had a spherical shape. This observation, coupled to the DLS results listed above, suggested that under these conditions, molecule 2 form aggregates of 100 nm supramolecular assemblies that act as a template for the crystal nucleation and growth. This outcome has been reported for liposomes and other spherical particles that efficiently sculpture calcite crystals.30 The above observations are in agreement with the FTIR spectra of the samples (Fig. S6†). The intensity of the absorption bands due to molecule 2 increases with its concentration in solution with respect to those due to CaCO3. Unfortunately, as observed for molecule 1, the co-presence of calcite crystals and of an additional molecule 2 phase does not allow quantification of molecule 2 entrapped in the crystals.
The influence of molecule 2 on the precipitation of CaCO3 was also studied using a 100 mM CaCl2 solution. The increase in Ca2+ concentration caused a higher starting CaCO3 supersaturation and a decrease in the Ca2+/molecule 2 molar ratio. Fig. S7† shows SEM images of the crystals obtained in the presence of different concentrations of molecule 2. At a concentration of 1 mM (Fig. S7A–C†), rhombohedral crystals formed, which sometimes aggregated (Fig. S4C†). Under these conditions, it seems that molecule 2 does not interact with crystals (Ca2+/molecule 2 molar ratio = 100). With the increase of the concentration of molecule 2 (10 mM, Ca2+/molecule 2 molar ratio = 10), calcite crystals showed {hk.0} faces together with the rhombohedral {10.4} faces. This observation suggested that the capability of molecule 2 to act as a crystal growth modifier is also affected by the starting conditions of supersaturation.
A clear view on the effect of the foldamers, molecule 1 and molecule 2, on the precipitation of CaCO3 is shown in Table 1. These molecules have similar structures and the same ionizable carboxylic groups; their assembly changes as a function of the chemistry of functional groups proximal to the carboxylic ones. In the case of molecule 1, the assembly state is a function of the concentration. Molecules 1 and 2 should interact with the crystallizing calcite in their assembled form since their assembly was studied with the Ca2+ concentration and pH used for CaCO3 precipitation. Thus, it is their physical state that governs the interactions. The diverse physical states should bring a different space distribution of the interacting ionizable carboxylic groups. As a consequence, a diverse interaction with CaCO3 is expected.31 This concept was deeply investigated for example by using self-assembled monolayers32 and liposomes33 as foldamer counterparts.15–17 The reported observations also agree with the behaviour of some biomineralization proteins that are assumed to adopt an ordered conformation and the geometry of ionizable functional groups to interact with biominerals.34,35
| Conc (mM) | Molecule 1 | Molecule 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| CaCO3 precipitation | CaCO3 precipitation | |||||||
| Ca2+/mol. molar ratio | Physical state | Polymorph/size (μm) | Shape/morphology | Ca2+/mol. molar ratio | Physical state | Polymorph/size (μm) | Shape/morphology | |
| 0 | — | — | Calcite/40 | Rhomb./{10.4} | — | — | Calcite/40 | Rhomb./{10.4} |
| 1 | 10 | Solution | Calcite/40 | Rhomb./{10.4} | 10 | Supram. aggregates | Calcite/40 | Rhomb./{10.4} |
| 5 | 2 | Highly viscous sol | Calcite/30 | Rhomb./{10.4}–{hk.0} | 2 | Supram. aggregates | Calcite/40 | Rhomb./{10.4}/cavities |
| 10 | 1 | Gel | Calcite/50 | Rhomb./{10.4}–{hk.0} | 1.0 | Supram. aggregates | Calcite/20 | Rhomb./{10.4}–{hk.0}/cavities |
Experimental
The synthesis of molecules 1 and 2 was carried out according to the literature reported in ref. 24. Shortly, they have been prepared from Boc-L-Phe-D-Oxd-OBn and Boc-L-Phe-D-Pro-OBn, respectively, that were deprotected with trifluoroacetic acid in dichloromethane to obtain in quantitative yield the corresponding trifluoroacetate salt, that in turn was coupled with azelaic acid using HBTU (O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate) and Et3N (triethylamine) as coupling agents. After purification by flash chromatography, both have been obtained in high yield. The benzyl esters have been transformed into the corresponding acids by hydrogenolysis, both in quantitative yield.The propensity of molecules 1 and 2 to form hydrogels was checked. The general method adopted to form gels was to place one compound in a small test tube (8 mm in diameter) and dissolve it in a pure solvent or in a solvent mixture. As ultrasounds influence the aggregation properties of the molecules in the solvents,36 the tube was sonicated for 20 minutes at room temperature, and then it was left to stand one night before checking the gel formation. As both molecules are not soluble in water, a 9:1 (v/v) water–ethanol mixture was used.
Dynamic light scattering was used for determination of the self-assembled molecule 2 size distributions employing a Malvern Nano ZS instrument with a 633 nm laser diode. Experiments were carried at 25 °C in a quartz cuvette with 1 cm optical path length.
A 30 cm × 30 cm × 50 cm crystallization chamber was used for the crystallization experiments of calcium carbonate. Two 25 mL beakers each containing about 20 g of (NH4)2CO3(s) (Carlo Erba), the source of NH3(g) and CO2(g) vapours, and two Petri dishes (d = 8 cm), each containing about 10 g of anhydrous CaCl2(s) (Fluka), a hygroscopic agent that controls humidity, were placed inside the chamber. Microplates for cellular culture (Microplate 24 well with Lid, IWAKI) containing a round glass cover slip in each well were used. Into each well, 750 μL of a 10 mM (or 100) CaCl2 solution in a 9/1 (v/v) H2O–EtOH mixture were poured. The additive, molecule 1 or molecule 2, was dissolved in the CaCl2 solution to obtain a final concentration equal to 1 mM, 5 mM or 10 mM. The microplate was covered with aluminum foil and a hole was made over every well. After 4 days, the obtained crystals were washed three times with the 9/1 (v/v) H2O–EtOH mixture, three times with milli-Q water (with a resistivity of 18.2 MΩ cm at 25 °C, filtered through a 0.22 μm membrane) and one time with ethanol; then they were air-dried and analysed. All of the experiments were conducted at room temperature. Each crystallization trial was replicated at least three times.
A Leica transmission optical microscope was used to obtain images of CaCO3 crystals. A microscope slide containing the sample was placed beneath a standard glass cover slip and observed under bright-field conditions with crossed polarizers. Images were captured with a CCD digital camera and recorded using the software (LAS EZ) supplied by Leica Microsystems. Image analysis was conducted on the crossed polarized optical micrographs to obtain info on crystallite birefringence. The dried glass cover slips covered with crystals were glued to the SEM aluminum stubs. Some samples were inspected with a Phenom™ scanning electron microscope. In addition, scanning electron micrographs of gold-sputtered samples were also recorded using a Hitachi 6400 field emission gun scanning electron microscope. The X-ray powder diffraction patterns were obtained using a Philips X'PertPro diffractometer. The diffraction patterns were collected using a voltage of 40 kV and a current of 40 mA. A diffraction region between 20° and 60° of 2θ was scanned. The measurements were carried out directly on the microscope slide on which particles were deposited. Infrared spectra were collected by using a Thermo Nicolet Avatar 370 FTIR spectrometer with KBr pellets, working in the range of 4000–400 cm−1, at a spectral resolution of 4 cm−1 and 64 scans for each acquisition.
Conclusions
This research adds new knowledge in the use of pseudopeptide foldamers to act as effective crystal growth modifiers. It shows that their self-assembly can be tuned by applying minor structural changes on the molecular backbone and that, as consequence of this, their capability to work as crystal growth modifiers changes. These observations are of general interest for the design of new molecules affecting the crystallization process and have implications in understanding how biological molecules control the growth of mineral phases.Acknowledgements
GF thanks the Consorzio Interuniversitario di Ricerca della Chimica dei Metalli nei Sistemi Biologici (CIRC MSB) for the support. CT thanks the Italian “Ministero dell'Istruzione, dell'Università e della Ricerca” (MIUR) (program PRIN 2010NRREPL_009) and “Consorzio CINMPIS” for the financial support. LM thanks the “Consorzio Spinner Regione Emilia Romagna” for the financial support.Notes and references
- S. Mann, Biomineralization: Principles and Concepts in Bioinorganic Materials, Chemistry, Oxford University Press, New York, 2001 Search PubMed.
- G. Falini and S. Fermani, Cryst. Res. Technol., 2013, 48, 864–876 CrossRef CAS.
- L. Xiang, Y. Wen, Q. Wang and Y. Jin, Mater. Chem. Phys., 2006, 98, 236–240 CrossRef CAS PubMed.
- L. Brecevic and D. Kralj, Croat. Chem. Acta, 2007, 80, 467–484 CAS.
- (a) J. J. De Yoreo and P. G. Vekilov in Biomineralization ed. P. M. Dove, J. J. De Yoreo and S. Weiner, Mineral Soc. Am., Washington, DC, 2003, pp. 57–93 Search PubMed; (b) H. Coelfen, Curr. Opin. Colloid Interface Sci., 2003, 8, 23–31 CrossRef.
- M. Gungormus, H. Fong, W. Kim, J. Spencer Evans, C. Tamerler and M. Sarikaya, Biomacromolecules, 2008, 9, 966–973 CrossRef CAS PubMed.
- G. Falini, M. Gazzano and A. Ripamonti, J. Mater. Chem., 2000, 10, 535–538 RSC.
- J. H. Harding, C. L. Freeman and D. M. Duffy, CrystEngComm, 2014, 16, 1430–1438 RSC.
- E. D. Sone, E. R. Zubarev and S. I. Stupp, Small, 2005, 1, 694–697 CrossRef CAS PubMed.
- S. B. Mukkamala and A. K. Powell, Chem. Commun., 2004, 918–919 RSC.
- S. Weiner and L. Addadi, Trends Biochem. Sci., 1993, 31, 7119–7126 Search PubMed.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4011 CrossRef CAS PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- J. Becerril, J. M. Rodriguez, H. Soraogi and A. D. Hamilton, in Foldamers: Structure, Properties, and Applications ed. S. Hecht and I. Huc, Wiley-VCH, Weinheim, 2007, pp. 217–228 Search PubMed.
- J. J. M. Donners, R. J. M. Nolte and N. A. J. M. Sommerdijik, J. Am. Chem. Soc., 2002, 124, 9700–9701 CrossRef CAS PubMed.
- N. Ueyama, H. Kozuki, M. Doi, Y. Yamada, K. Takahashi, A. Onoda, T. Hokamura and H. Yamamoto, Macromolecules, 2001, 124, 9700–9701 Search PubMed.
- L. A. Estroff, C. D. Incarvito and A. D. Hamilton, J. Am. Chem. Soc., 2004, 126, 2–3 CrossRef CAS PubMed.
- C. Tomasini, G. Angelici and N. Castellucci, Eur. J. Org. Chem., 2011, 3648–3669 CrossRef CAS.
- (a) C. Tomasini and M. Villa, Tetrahedron Lett., 2001, 42, 5211–5214 CrossRef CAS; (b) F. Bernardi, M. Garavelli, M. Scatizzi, C. Tomasini, V. Trigari, M. Crisma, F. Formaggio, C. Peggion and C. Toniolo, Chem. – Eur. J., 2002, 8, 2516–2525 CrossRef.
- (a) G. Angelici, G. Falini, H.-J. Hofmann, D. Huster, M. Monari and C. Tomasini, Angew. Chem., Int. Ed., 2008, 47, 8075–8078 CrossRef PubMed; (b) G. Angelici, G. Falini, H.-J. Hofmann, D. Huster, M. Monari and C. Tomasini, Chem. – Eur. J., 2009, 15, 8037–8048 CrossRef CAS PubMed.
- N. Castellucci, G. Sartor, N. Calonghi, C. Parolin, G. Falini and C. Tomasini, Beilstein J. Org. Chem., 2013, 9, 417–424 CrossRef CAS PubMed.
- (a) J. Becerril, M. I. Burguete, B. Escuder, F. Galindo, R. Gavara, J. F. Miravet, S. V. Luis and G. Peris, Chem. – Eur. J., 2004, 10, 3879–3890 CrossRef CAS PubMed; (b) J. Becerril, B. Escuder, J. F. Miravet, R. Gavara and S. V. Luis, Eur. J. Org. Chem., 2005, 481–485 CrossRef CAS.
- (a) T. Shimizu, Macromol. Rapid Commun., 2002, 23, 311–331 CrossRef CAS; (b) J.-H. Fuhrhop and T. Wang, Chem. Rev., 2004, 104, 2901–2937 CrossRef CAS PubMed.
- N. Castellucci, G. Angelici, G. Falini, M. Monari and C. Tomasini Eur, J. Org. Chem., 2011, 3082–3088 Search PubMed.
- N. Castellucci, G. Sartor, N. Calonghi, C. Parolin, G. Falini and C. Tomasini Beil, J. Org. Chem., 2013, 9, 417–424 CAS.
- J. Ihli, P. Bots, A. Kulak, L. G. Benning and F. C. Meldrum, Adv. Funct. Mater., 2013, 23, 1965–1973 CrossRef CAS.
- (a) G. B. Ramírez-Rodríguez, J. M. Delgado-López and J. Gómez-Morales, CrystEngComm, 2013, 15, 2206–2212 RSC; (b) J. Gómez-Morales, J. M. Delgado-López, M. Iafisco, A. Hernández-Hernández and M. Prat, Cryst. Growth Des., 2011, 11, 4802–4809 CrossRef.
- G. Falini, M. Gazzano and A. Ripamonti, Chem. Commun., 1996, 1037–1038 RSC.
- H. Y. Li, H. L. Xin, D. A. Muller and L. A. Estroff, Science, 2009, 326, 1244–1247 CrossRef CAS PubMed.
- (a) P. Wan, Y. Zhao, H. Tong, Z. Yang, Z. Zhu, X. Shen and J. Hu, Mater. Sci. Eng., C, 2009, 29, 222–227 CrossRef CAS PubMed; (b) A. Szczes, Colloids Surf., B, 2013, 101, 44–48 CrossRef CAS PubMed.
- L. Addadi and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4110–4114 CrossRef CAS.
- Y.-J. Han and J. Aizenberg, Angew. Chem., Int. Ed., 2003, 42, 3668–3670 CrossRef CAS PubMed.
- C. C. Tester and D. Joester, Methods Enzymol., 2013, 532, 257–276 CAS.
- J. S. Evans, Chem. Rev., 2008, 108, 4455–4462 CrossRef CAS PubMed.
- A. Adamiano, S. Bonacchi, N. Calonghi, D. Fabbri, G. Falini, S. Fermani, D. Genovese, D. Kralj, M. Montalti, B. Njegić Džakula, L. Prodi and G. Sartor, Chem. – Eur. J., 2012, 18, 14367–14374 CrossRef PubMed.
- (a) Y. Y. Choi, Y. Jeong, M. K. Joo and B. Jeong, Macromol. Biosci., 2009, 9, 869–874 CrossRef PubMed; (b) Y. Y. Choi, M. K. Joo, Y. S. Sohn and B. Jeong, Soft Matter, 2008, 4, 2383–2387 RSC; (c) K. Isozaki, H. Takaya and T. Naota, Angew. Chem., Int. Ed., 2007, 46, 2855–2857 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DLS profiles, XRD patterns, and SEM pictures of calcium carbonate crystals. See DOI: 10.1039/c4ce01569j |
| This journal is © The Royal Society of Chemistry 2015 |