
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reliable palladium nanoparticle syntheses in aqueous solution: the importance of understanding precursor chemistry and growth mechanism†
Frieder
Kettemann
a,
Maria
Wuithschick
a,
Gianvito
Caputo
a,
Ralph
Kraehnert
b,
Nicola
Pinna
a,
Klaus
Rademann
a and
Jörg
Polte
*a
aDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Strasse 2, 12489 Berlin, Germany. E-mail: joerg.polte@chemie.hu-berlin.de
bTechnische Chemie, Technische Universität Berlin, Strasse des 17 Juni 124, 10623 Berlin, Germany
First published on 13th January 2015
Abstract
Reliable protocols for the synthesis of palladium nanoparticles (Pd-NPs) in aqueous solution are rarely found and the corresponding growth mechanisms often remain unknown. Furthermore, syntheses of Pd-NPs always demand the use of stabilizing agents which are often unfavorable for catalytic applications. In this contribution, the importance of the palladium precursor chemistry as a prerequisite for any reliable Pd-NP synthesis in aqueous solution is shown. This includes a detailed study of the influence of the precursor chemistry on the nanoparticle growth mechanism. The findings enable the controlled modification of a common synthetic protocol (i.e. the reduction of a palladium precursor with NaBH4) to obtain sub-5 nm Pd-NPs without the use of any stabilizing agent. In addition, it is also shown that such mechanistic studies are not only of great importance to the development of novel synthetic procedures. Exemplarily, the successful transfer of the synthesis from lab- to large-scale is demonstrated.
Introduction
Noble metal nanoparticles (NPs) are widely studied due to their manifold applications in electronics, medicine and catalysis.1–3 In particular, palladium nanoparticles (Pd-NPs) are of great scientific and commercial interest because of their unique catalytic properties.4–6The catalytic performance of Pd-NPs strongly depends on their size (in particular in the sub-10 nm regime), shape, and surface modification (i.e. surfactants, ligands, and coordinating solvents).7–15 Only few reliable Pd-NP syntheses are described in the literature which in general demand the use of stabilizing agents (i.e. surface modification).14 In addition, the syntheses are often carried out in organic solvents that are unfavorable for many applications.10,16 Surface modification can decrease the catalytic activity of NPs by blocking their active sites. In addition, stabilizing agents can complicate the further use of NPs in catalyst syntheses (e.g. immobilization of NPs in porous layers).
Consequently, reliable synthetic protocols for NPs with a defined size in aqueous solution without any stabilizing agent are highly demanded.14,17,18 These solely electrostatically stabilized NPs are free of organic stabilizers or ligands and therefore herein called “naked”. Such “naked” NPs can be used directly for catalytic applications, both in aqueous solution and for solid supports.17,19,20 Furthermore, “naked” Pd-NPs can be functionalized with ligands according to the desired application (e.g. chiral ligands for enantioselective catalysis21–23), without the elaborate removal or exchange of surfactants.14,18 “Naked” Pd-NPs can also serve as a reference system for the catalytic properties of NPs.18
This contribution addresses: (i) general issues in reliable Pd-NP syntheses in aqueous solution, in particular the precursor chemistry; and (ii) the mechanistic understanding of NP growth. The results enabled the design of a reliable synthetic procedure for “naked” sub-5 nm Pd-NPs in aqueous solution based on the reduction of a metal precursor with NaBH4. Furthermore, it is also shown that such mechanistic studies are not only of great importance to the development or improvement of synthetic procedures. Exemplarily, crucial points in the successful upscaling of a lab-scale procedure are deduced for this novel synthesis.
Experimental section
All chemicals were used as purchased without any purification. Ultrapure water (18.2 MΩ cm) was used as a solvent for all reactions.Synthesis of Pd-NPs without pH adjustment: 5 ml of a freshly prepared 3 mM NaBH4 (Sigma Aldrich, 99.99%) solution were mixed in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with 5 ml of a 0.5 mM solution of K2PdCl4 (Alfa Aesar, 99.999%) using Eppendorf pipettes.
:1 with 5 ml of a 0.5 mM solution of K2PdCl4 (Alfa Aesar, 99.999%) using Eppendorf pipettes.
Synthesis of Pd-NPs with pH adjustment: the pH value of the precursor solution was adjusted by addition of NaOH (Carl Roth, p.a.) or HClO4 (Sigma Aldrich, 99.99%). The synthesis was carried out as described above.
Characterization: UV-vis spectra of the precursor solutions were measured with a SEC2000 spectrometer (BAS Inc.). SAXS experiments were carried out with a lab-scale setup (SAXSess, Anton Paar) using a flow through quartz cuvette. The SAXS data were evaluated assuming spherical particles with a Schulz–Zimm distribution. Details of the mathematical modeling24 and the experimental setups can be found in the ESI.†
Results and discussion
This contribution consists of four parts. First, the chemistry of the Pd precursors in water is discussed as a key issue in the controlled syntheses of Pd-NPs. In the second part, the growth mechanism of the Pd-NPs synthesized via the reduction of K2PdCl4 with NaBH4 is investigated. In the third part, the influence of the precursor chemistry on the NP growth is discussed and a synthetic procedure for “naked” Pd-NPs is deduced. Finally, the upscaling of the lab-scale NP synthesis is demonstrated.Pd hydrolysis in aqueous solution
Palladium salts such as alkali tetrachloropalladates (X2PdCl4) or palladium nitrate (Pd(NO3)2) are typically used as precursors for Pd-NP syntheses.10,13,14,25–27 In aqueous solution, palladium ions form aqua complexes that are subject to pH dependent hydrolysis.28 Subsequent olation processes result in the formation of polynuclear hydroxo complexes (herein denoted as PHC):29| [Pd(H2O)4]2+ → [Pd(OH)4]2− + 4H+ |
| n[Pd(OH)4]2− → [Pd(OH)2]n + 2n OH− |
Although hydrolysis of Pd complexes is the focus of several publications,29–34 its influence on the syntheses of Pd-NPs in aqueous solution is to the best of our knowledge not described in the literature. In this section the hydrolysis of Pd complexes in aqueous solution and its influence on Pd-NP syntheses are discussed exemplarily for K2PdCl4. A detailed study that includes other common Pd precursors can be found in S2 in the ESI.†
Fresh solutions of K2PdCl4 in a typical concentration for NP syntheses (e.g. 0.25–3 mM) are transparent and yellow. Within minutes, the color of the precursor solutions changes to brown due to the formation of PHC. The PHC precipitate within hours (see Fig. 1a). This process of precursor aging was investigated with time-resolved UV-vis and small angle X-ray scattering (SAXS). Selected UV-vis spectra and scattering curves of the precursor solution during the first hour of aging are displayed in Fig. 1b and c. The UV-vis spectrum of a fresh K2PdCl4 solution shows absorption maxima at 310 nm and 420 nm that are typical of ligand-to-metal charge transfer bands of [PdCl3H2O]− in water.32,35 Within seconds, a broad background absorbance evolves with increasing intensities towards shorter wavelengths which most likely indicates the formation of PHC.32 After several hours, the absorbance decreases caused by PHC precipitation (for details see S2 in the ESI†).
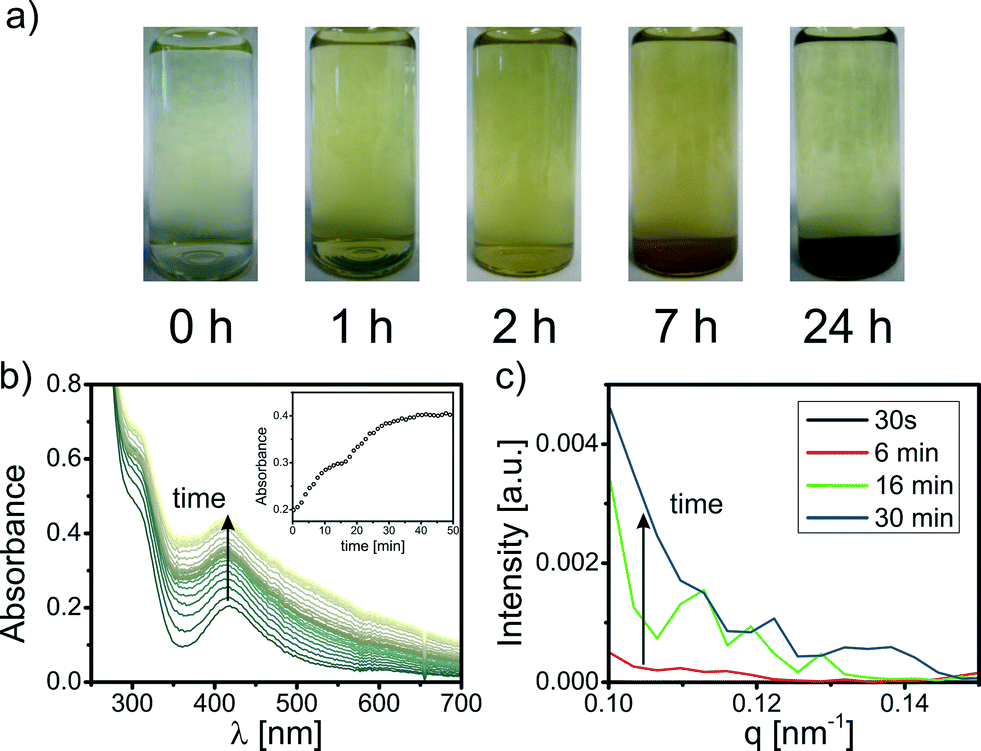 | ||
| Fig. 1 (a) Digital photographs of the aging process of an aqueous K2PdCl4 solution (1 mM). (b) Time-resolved UV-vis measurements of 1 h aging of a 1 mM K2PdCl4 solution (spectra were recorded at 1 min intervals); the inset shows the absorbance at 420 nm as a function of time. (c) Scattering curves of a 0.5 mM K2PdCl4 solution in linear representation. | ||
The scattering curves of a K2PdCl4 solution at different aging times are displayed in Fig. 1c. Scattering of PHC is not observed for a fresh Pd precursor solution (aging time less than 30 s). At aging times greater than 2 min, the intensity increases at low q values, indicating the formation of larger colloidal PHC (for details see S2.4 in the ESI†). The PHC size can be estimated to be larger than 20 nm in radius. Upon aging of the Pd solution, PHC grow further until precipitation. Scattering curves and UV-vis data for the aging of Na2PdCl4 and Pd(NO3)2 solutions can be found in Fig. S3–S6 in the ESI.† In general, they show the same characteristics.
The precipitation of PHC strongly depends on the presence of Cl− ions. In Pd solutions without Cl− (e.g. aqueous Pd(NO3)2 solutions), the formed PHC do not precipitate within weeks. The addition of small amounts of Cl− to fresh Pd(NO3)2 solution results in precipitation upon few hours of aging as is known for palladates.
The time-resolved UV-vis and SAXS measurements of the precursor aging show that Pd salts start to hydrolyze within seconds. This fast formation of PHC has to be considered for Pd-NP syntheses in aqueous solution. Thus, precursor solutions have to be used immediately after preparation (aging time: <10 s) for Pd-NP synthesis to avoid the formation of relevant amounts of PHC. At this extremely short aging time, formation of PHC is not observed by UV-vis and SAXS. Hence, in very fresh precursor solutions the formation of PHC can be neglected.
Mechanistic studies
The synthesis of Pd-NPs is the focus of many publications, yet the underlying mechanisms of particle formation are rarely investigated.36 The investigation of the NP growth mechanism and parameters that determine the particle size is an essential step towards a directed nanoparticle design.37Previously, we deduced the growth mechanisms of gold and silver nanoparticles.37–40 In a recent publication, we showed that the reduction of gold and silver precursors by NaBH4 is a very fast process which occurs within the mixing time of the precursor and reducing agent solution. Thus, the reduction is much faster than the actual particle growth.38 It was shown that the growth mechanism of gold NPs consists of one coalescence step whereby the corresponding silver NP synthesis comprises two well separated steps of coalescence.
For the synthesis of Pd-NPs, a fresh 0.5 mM aqueous solution of K2PdCl4 was mixed in a ratio of 1:1 with a 3 mM solution of NaBH4 (for details see S1 in the ESI†). Since NaBH4 hydrolyzes in aqueous solution, it was freshly prepared and used within approx. 10 min.41 Upon mixing of K2PdCl4 and NaBH4, the solution turned immediately from yellow to light brown, indicating the formation of Pd-NPs. After approx. 10–20 min, a color change from brown to gray-black was observed.
The particle growth mechanism was investigated using time-resolved SAXS measurements. The first few seconds of the particle growth were measured using a continuous flow setup (CFS). Further growth was monitored with a standard SAXS setup.
The CFS was employed to overcome the limited time-resolution of lab-scale SAXS setups. A detailed description of the CFS can be found in the ESI† (S6.1) and in a previous publication.38 Briefly, a CFS transforms the time scale into a length scale. A syringe pump was used to mix the reactants via Teflon tubing and mixers. The adjustment of the tube length and flow rate enables millisecond time resolution. The samples were measured using a standard SAXS flow through quartz capillary.
The results of the CFS experiments are displayed in Fig. 2a and b. Within the first few seconds of the synthesis, the Pd-NPs grow from 0.8 to 1.2 nm in radius (polydispersity of 20%). The volume fraction remains rather constant throughout the synthesis, while the relative number of particles decreases, indicating growth by coalescence.38
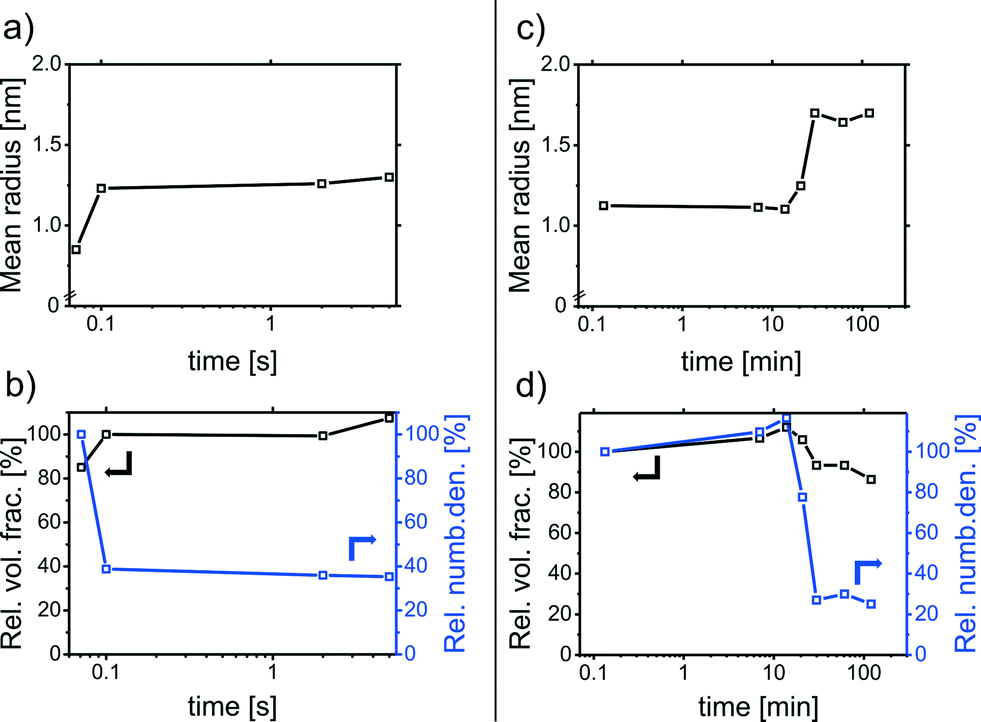 | ||
| Fig. 2 Results of time-resolved SAXS measurements for the investigation of the particle growth mechanism: (a) the mean particle radius (polydispersity, 20%) and (b) relative volume fraction and relative number density for the first few milliseconds of particle growth derived from the continuous flow setup (CFS) measurements; (c) and (d) results of the further particle growth within 2 h (polydispersity, 25%). | ||
Further particle growth was investigated by extracting samples from a stirred NP solution which were subsequently measured with a standard lab-scale SAXS setup (time resolution was in a range of about 1 min). A PVP solution was added to the samples to prevent NP aggregation during the measurement. Fig. 2c and d show the SAXS results for reaction times up to 2 h. The first data point corresponds to a sample extracted after 8 s and reveals particles with a mean radius of approx. 1.1 nm. This is in accordance with the particles measured with the CFS after 5 s. After about 20 min, a further particle growth step is observed. At this point the color of the reaction solution changed from light brown to gray-black. The final particles have a mean radius of approx. 1.8 nm (polydispersity, 25%).
The growth is due to coalescence since the volume fraction remains rather constant throughout the synthesis. The results are similar to the corresponding Ag-NPs synthesized by the reduction of AgClO4 with aqueous NaBH4.37,40 As a result, the particle growth and final size are only governed by colloidal stability.38,40 The deduced growth mechanism is displayed schematically in Fig. 3. The particle growth consists of two distinct steps of coalescence separated by a metastable phase. The metastable particles are formed within seconds by the coalescence of Pd clusters. After 10–20 min, the metastable particles grow in a second coalescence step to give the final particles.
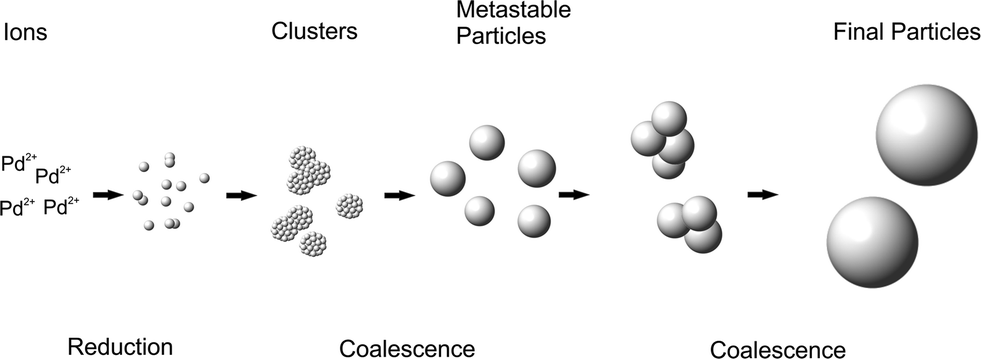 | ||
| Fig. 3 Schematic illustration of the deduced growth mechanism of Pd-NPs. | ||
The second coalescence is a consequence of an abrupt decrease of colloidal stability. We assume that the decrease of colloidal stability coincides with the full consumption of NaBH4 as observed for Ag-NPs.37 Pd-NPs were shown to form an oxide layer in aqueous solution42,43 that can decrease the colloidal stability of the Pd-NPs. Excess BH4− in the reaction mixture could continuously reverse such surface oxidation. With the full consumption of BH4−, an oxide layer is formed on the surface, decreasing the colloidal stability. As a consequence, growth due to coalescence is induced.
The final Pd-NPs are not long-term stable and they precipitate within days. The particles should be used within 1–2 h after the synthesis or stabilized with a stabilizing agent to prevent aggregation. The stabilizing agent can be selected according to the required application.
Influence of precursor aging on NP growth
In a previous section it was shown that the formation of PHC is a fast process in aqueous Pd precursor solutions. To evaluate the influence of PHC on NP formation, the Pd-NP synthesis was investigated using an aged precursor solution (6 min). SAXS measurements of the final particles revealed a broad NP size distribution with mean radii between 2 nm and 20 nm.An aged Pd precursor solution contains two Pd species in unknown concentrations: dissolved Pd2+ ions and aggregated PHC. The growth mechanism of NPs formed from the dissolved Pd2+ in an aged solution is most likely not influenced by the presence of PHC. Particles with radii of approx. 2 nm are also formed in the aged solutions. Furthermore, the characteristic color change from light brown to gray-black after approx. 20 min indicates that the second growth step still occurs.
The large particles most likely result from the reduction of PHC since the PHC in aged precursor solutions are in the same size range as the large Pd particles in the NP solution. Thus, the broad size distribution originates from the simultaneous reduction of dissolved Pd2+ and PHC leading to two different mechanisms of particle formation. The large particles originate from the reduction of PHC. In addition, the reduction of dissolved Pd2+ leads to small particles as described in the previous section and as depicted in Fig. 4.
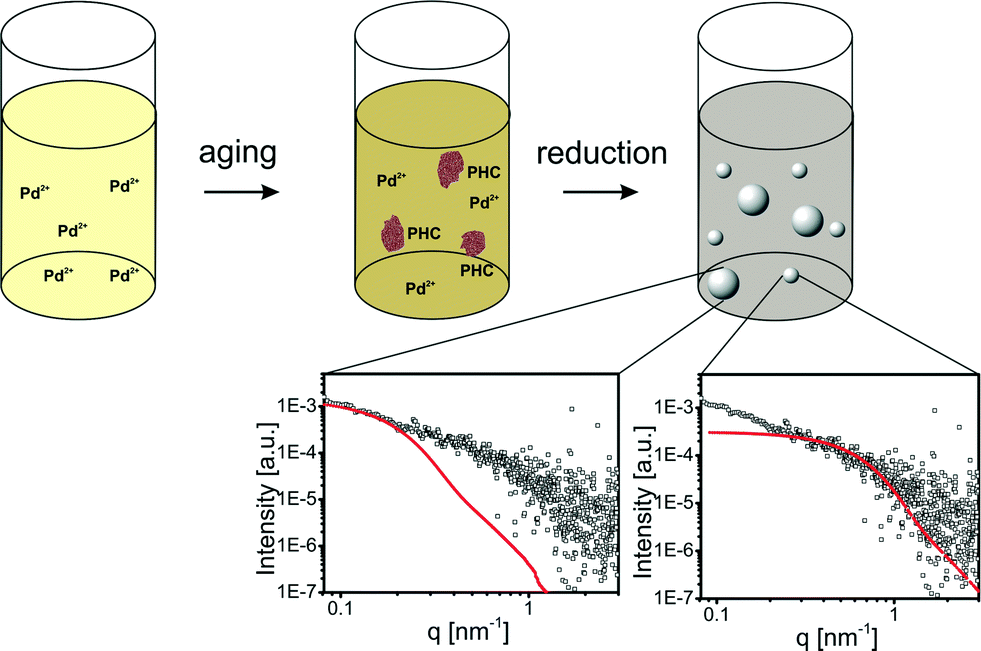 | ||
| Fig. 4 Schematic illustration of the Pd-NP synthesis with aged precursors; aged precursor solutions contain Pd2+ and polynuclear hydroxo complexes (PHC). The chemical reduction of an aged Pd precursor solution leads to NPs with a broad size distribution; large particles originate from the reduction of PHC and cause scattering at low q values, while small particles derive from the reduction of dissolved Pd2+ and cause scattering at high q values. | ||
Precursor stabilization and upscaling
It was shown that the hydrolysis of palladium salts and the formation of PHC are crucial issues in the synthesis of monodisperse sub-10 nm Pd-NPs in aqueous solution. The formation of relevant amounts of PHC in precursor solutions can be avoided by using very fresh precursor solutions (aging time: <10 s). However, this is not practical and often not possible. Thus, the hydrolysis of Pd complexes is a general issue in the synthesis of Pd-NPs that needs to be considered if the precursor solution cannot be used within a few seconds. This applies in particular to large-scale syntheses, seed-mediated growth procedures, synthetic procedures at elevated temperatures (which accelerate the hydrolysis44) and syntheses using mild reducing agents.The stabilization of precursor solutions is necessary in order to overcome the issue of precursor instability. The formation of PHC can be delayed or inhibited at low pH values.34,45 However, low pH values involve high ionic strengths which decrease the colloidal stability of NPs. As a result, NPs synthesized with strongly acidic precursor solutions (pH < 2) precipitate immediately if no further stabilizing agents are used. Thus, very acidic precursor solutions are inappropriate for synthesis of sub-5 nm and in particular “naked” Pd-NPs.
Consequently, for the synthesis of “naked” sub-5 nm Pd-NPs, a balance between precursor and colloidal stability is required. For the above described synthetic procedure, such a balance was found at a pH value of 3.3 (pH value was adjusted with HClO4). Formation of PHC at a pH value of 3.3 in the precursor solution is not observed for at least 30 min (scattering curves are displayed in Fig. S5 in the ESI†). From a precursor solution with a pH value of 3.3, stable particles with a mean radius of 2.3 nm (polydispersity, 30%) are reproducibly formed without the addition of steric stabilizers as revealed from SAXS measurements. The particles formed from pH-stabilized precursors are only slightly larger than those synthesized with fresh and non-acidic Pd precursor solutions (for details see S3 in the ESI†). In accordance with the SAXS experiments, TEM images of the final particles show spherical NPs with a mean radius of 2.2 nm (see S4 in the ESI†).
As shown, pH-stabilization of precursor solutions is necessary for several synthetic procedures. The relevance of precursor stabilization is illustrated by upscaling the Pd-NP synthesis developed beforehand. This demonstrates a principal synthetic procedure for a large-scale synthesis.
The Pd-NP synthesis was upscaled to 500 ml by the reduction of 250 ml of K2PdCl4 (0.5 mM) with 250 ml of NaBH4 (3 mM) using a peristaltic pump, a Teflon mixer and Teflon tubing (for details see S5 in the ESI†). Note that the reactant volumes can easily be increased with appropriate pumping and mixing systems. The synthesis was conducted with and without pH-stabilized precursor solutions whereby the duration of mixing was varied at 1 and 10 min by adjusting the flow rates. The precursor solutions were always freshly prepared. Thus, for the mixing times of 1 and 10 min the precursor solution was aged between 0–1 min and 0–10 min, respectively. At the relatively short mixing time of 1 min, SAXS measurements reveal final Pd-NPs with radii of 2.2 nm with a broad size distribution (polydispersity, ~50%, see Fig. S10 in the ESI†) if a non-pH-stabilized precursor solution is used. Increasing the mixing time to 10 min results in NPs with a bimodal size distribution, typical of NPs synthesized with aged precursor solutions.
If a precursor solution with a pH value of 3.3 is used for the upscaled synthesis, the formation of PHC is inhibited and the particle size and size distribution are not dependent on the mixing duration. Particles with a mean radius of 2.1 nm and a polydispersity of 25% are reproducibly formed for mixing durations of 1 and 10 min. As already stated, the addition of a stabilizing agent is necessary, approx. within 1–2 h after the synthesis, to assure long-term stability of the as-prepared particles.
The comparison of the final particle sizes from the lab-scale and the upscaled synthesis using pH-stabilized precursors reveals the dependence of the particle size distribution on the mixing conditions. It was found that uniform mixing (i.e. applying a pump and Teflon mixer) leads to slightly smaller NPs with a lower size distribution than less uniform mixing (i.e. using simple Eppendorf pipettes). The influence of the mixing conditions on the final particle size is described in the ESI† (see S7).
Hence, a precursor solution with a pH of 3.3 enables the reproducible large-scale synthesis of Pd-NPs with a mean radius of 2.1 nm.
Conclusions
This contribution addresses major aspects that have to be considered in the synthesis of Pd-NPs in aqueous solution and investigates the growth mechanism of Pd-NPs. An approach to the synthesis of “naked” Pd-NPs is presented based on the reduction of aqueous K2PdCl4 with NaBH4 which demands either fresh (aging: <10 s) or pH-stabilized precursor solutions.The hydrolysis of Pd complexes and the formation of PHC could be identified as crucial issues prohibiting reproducible synthesis of Pd-NPs with low polydispersities in aqueous solutions. The formation of PHC in the precursor solution can easily be detected prior to use by UV-vis spectroscopy through increasing background absorbance.
To avoid the formation of PHC, precursor solutions have to be used immediately after preparation. If immediate use is not possible, PHC formation can be delayed or inhibited by lowering the pH value of the precursor solution. This process is particularly important to all synthetic procedures applying slow reducing agents and elevated temperatures and to seeded-growth or large-scale syntheses.
Furthermore, it was shown that the growth of Pd-NPs in the investigated system is only due to coalescence in two separated steps similar to that of the corresponding Ag-NPs.
Mechanistic understanding and the use of mildly acidic precursor solutions enabled the reliable synthesis of “naked” Pd-NPs (i.e. surfactant free Pd-NPs) in the sub-5 nm regime. The as-prepared NPs were stable for 1–2 hours and could be used directly in catalytic applications applied to solid supports or functionalized with the desired stabilizing agents.
Acknowledgements
J.P. acknowledges generous funding by the Deutsche Forschungsgemeinschaft under the project PO 1744/1-1. F.K. acknowledges support by the IMPRS “Functional Interfaces in Physics and Chemistry”. M.W. acknowledges financial support by the Fonds der Chemischen Industrie. RK acknowledges generous BMBF funding (ELYKAT, FKZ 03EK3009).Notes and references
- A. N. Shipway, E. Katz and I. Willner, ChemPhysChem, 2000, 1, 18–52 CrossRef CAS.
- L. Zhang, F. X. Gu, J. M. Chan, A. Z. Wang, R. S. Langer and O. C. Farokhzad, Clin. Pharmacol. Ther., 2007, 83, 761–769 CrossRef PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- R. Narayanan and M. A. El-Sayed, J. Catal., 2005, 234, 348–355 CrossRef CAS PubMed.
- S. Cheong, J. D. Watt and R. D. Tilley, Nanoscale, 2010, 2, 2045–2053 RSC.
- O. M. Wilson, M. R. Knecht, J. C. Garcia-Martinez and R. M. Crooks, J. Am. Chem. Soc., 2006, 128, 4510–4511 CrossRef CAS PubMed.
- F. P. Zamborini, S. M. Gross and R. W. Murray, Langmuir, 2001, 17, 481–488 CrossRef CAS.
- S. U. Son, Y. Jang, K. Y. Yoon, E. Kang and T. Hyeon, Nano Lett., 2004, 4, 1147–1151 CrossRef CAS.
- R. Andrés, E. de Jesús and J. C. Flores, New J. Chem., 2007, 31, 1161–1191 RSC.
- Y. Piao, Y. Jang, M. Shokouhimehr, I. S. Lee and T. Hyeon, Small, 2007, 3, 255–260 CrossRef CAS PubMed.
- R. Narayanan and M. A. El-Sayed, Top. Catal., 2008, 47, 15–21 CrossRef CAS.
- E. Sadeghmoghaddam, C. Lam, D. Choi and Y.-S. Shon, J. Mater. Chem., 2010, 21, 307–312 RSC.
- M. Jin, H. Liu, H. Zhang, Z. Xie, J. Liu and Y. Xia, Nano Res., 2011, 4, 83–91 CrossRef CAS PubMed.
- N. Naresh, F. G. S. Wasim, B. P. Ladewig and M. Neergat, J. Mater. Chem. A, 2013, 1, 8553–8559 CAS.
- Q. Yuan, J. Zhuang and X. Wang, Chem. Commun., 2009, 6613–6615 RSC.
- B. Lim, M. Jiang, J. Tao, P. H. C. Camargo, Y. Zhu and Y. Xia, Adv. Funct. Mater., 2009, 19, 189–200 CrossRef CAS.
- S. Ikeda, S. Ishino, T. Harada, N. Okamoto, T. Sakata, H. Mori, S. Kuwabata, T. Torimoto and M. Matsumura, Angew. Chem., 2006, 118, 7221–7224 CrossRef.
- Z. Niu and Y. Li, Chem. Mater., 2014, 26, 72–83 CrossRef CAS.
- A. Guiet, T. Reier, N. Heidary, D. Felkel, B. Johnson, U. Vainio, H. Schlaad, Y. Aksu, M. Driess, P. Strasser, A. Thomas, J. Polte and A. Fischer, Chem. Mater., 2013, 25, 4645–4652 CrossRef CAS.
- E. Ortel, S. Sokolov, C. Zielke, I. Lauermann, S. Selve, K. Weh, B. Paul, J. Polte and R. Kraehnert, Chem. Mater., 2012, 24, 3828–3838 CrossRef CAS.
- M. Tamura and H. Fujihara, J. Am. Chem. Soc., 2003, 125, 15742–15743 CrossRef CAS PubMed.
- S. Jansat, M. Gómez, K. Philippot, G. Muller, E. Guiu, C. Claver, S. Castillón and B. Chaudret, J. Am. Chem. Soc., 2004, 126, 1592–1593 CrossRef CAS PubMed.
- K. Sawai, R. Tatumi, T. Nakahodo and H. Fujihara, Angew. Chem., Int. Ed., 2008, 47, 6917–6919 CrossRef CAS PubMed.
- M. Kotlarchyk, R. B. Stephens and J. S. Huang, J. Phys. Chem., 1988, 92, 1533–1538 CrossRef CAS.
- A. Nemamcha, J.-L. Rehspringer and D. Khatmi, J. Phys. Chem. B, 2006, 110, 383–387 CrossRef CAS PubMed.
- V. L. Nguyen, D. C. Nguyen, H. Hirata, M. Ohtaki, T. Hayakawa and M. Nogami, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2010, 1, 035012 CrossRef.
- G. Berhault, M. Bausach, L. Bisson, L. Becerra, C. Thomazeau and D. Uzio, J. Phys. Chem. C, 2007, 111, 5915–5925 CAS.
- C. F. Baes and R. S. Mesmer, Ber. Bunsen-Ges., 1977, 81, 245–246 Search PubMed.
- S. Y. Troitskii, A. L. Chuvilin, D. I. Kochubei, B. N. Novgorodov, V. N. Kolomiichuk and V. A. Likholobov, Russ. Chem. Bull., 1995, 44, 1822–1826 CrossRef.
- L. I. Elding and L.-F. Olsson, Inorg. Chim. Acta, 1986, 117, 9–16 CrossRef CAS.
- B. Didillon, E. Merlen, T. Pagès and D. Uzio, in Studies in Surface Science and Catalysis, ed. P. A. J. B. Delmon, Elsevier, 1998, vol. 118, pp. 41–54 Search PubMed.
- P. A. Simonov, S. Y. Troitskii and V. A. Likholobov, Kinet. Catal., 2000, 41, 255–269 CrossRef CAS.
- J.-F. Boily and T. M. Seward, Geochim. Cosmochim. Acta, 2005, 69, 3773–3789 CrossRef CAS PubMed.
- N. Torapava, L. I. Elding, H. Mändar, K. Roosalu and I. Persson, Dalton Trans., 2013, 42, 7755–7760 RSC.
- L. Rasmussen, C. K. Jørgensen, J. Sjövall and N. A. Zaidi, Acta Chem. Scand., 1968, 22, 2313–2323 CrossRef CAS PubMed.
- B. Lim, H. Kobayashi, P. H. C. Camargo, L. F. Allard, J. Liu and Y. Xia, Nano Res., 2010, 3, 180–188 CrossRef CAS PubMed.
- M. Wuithschick, B. Paul, R. Bienert, A. Sarfraz, U. Vainio, M. Sztucki, R. Kraehnert, P. Strasser, K. Rademann, F. Emmerling and J. Polte, Chem. Mater., 2013, 25, 4679–4689 CrossRef CAS.
- J. Polte, R. Erler, A. F. Thunemann, S. Sokolov, T. T. Ahner, K. Rademann, F. Emmerling and R. Kraehnert, ACS Nano, 2010, 4, 1076–1082 CrossRef CAS PubMed.
- J. Polte, T. T. Ahner, F. Delissen, S. Sokolov, F. Emmerling, A. F. Thuenemann and R. Kraehnert, J. Am. Chem. Soc., 2010, 132, 1296–1301 CrossRef CAS PubMed.
- J. Polte, X. Tuaev, M. Wuithschick, A. Fischer, A. F. Thuenemann, K. Rademann, R. Kraehnert and F. Emmerling, ACS Nano, 2012, 6, 5791–5802 CrossRef CAS PubMed.
- J. Andrieux, U. B. Demirci, J. Hannauer, C. Gervais, C. Goutaudier and P. Miele, Int. J. Hydrogen Energy, 2011, 36, 224–233 CrossRef CAS PubMed.
- L. M. Rossi, F. P. Silva, L. L. R. Vono, P. K. Kiyohara, E. L. Duarte, R. Itri, R. Landers and G. Machado, Green Chem., 2007, 9, 379–385 RSC.
- Y. Sun, L. Zhang, H. Zhou, Y. Zhu, E. Sutter, Y. Ji, M. H. Rafailovich and J. C. Sokolov, Chem. Mater., 2007, 19, 2065–2070 CrossRef CAS.
- L. A. Koroleva, N. D. Shikina, P. G. Kolodina, A. V. Zotov, B. R. Tagirov, Y. V. Shvarov, V. A. Volchenkova and Y. K. Shazzo, Geochem. Int., 2012, 50, 853–859 CrossRef CAS.
- T. Shi, L. I. Elding, M. Y. Meľnikov, V. I. Feldman, Á. G. Kristjánsdóttir, O. Matsson, K. V. Mikkelsen and A. Senning, Acta Chem. Scand., 1998, 52, 897–902 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional text and experimental data, study of precursor aging, including photographs, time resolved UV-vis and SAXS experiments, and XRD data; details of the synthesis of Pd nanoparticles; TEM images of Pd-NPs; experimental data for the large scale synthesis; a reproducibility study; experimental setups for the large scale synthesis and the CFS; study of the influence of mixing conditions; and evaluation of the SAXS data. See DOI: 10.1039/c4ce01025f |
| This journal is © The Royal Society of Chemistry 2015 |