
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective [4+4] photodimerization of anthracene-2,6-dicarboxylic acid mediated by a C2-symmetric chiral template†
Mark M.
Maturi
a,
Gaku
Fukuhara
b,
Koichiro
Tanaka
b,
Yuko
Kawanami
b,
Tadashi
Mori
b,
Yoshihisa
Inoue
*b and
Thorsten
Bach
*a
aDepartment Chemie and Catalysis Research Center (CRC), Technische Universität München, 85747 Garching, Germany. E-mail: thorsten.bach@ch.tum.de; Fax: +49 89 28913315; Tel: +49 89 28913330
bDepartment of Applied Chemistry, Osaka University, 2-1 Yamada-oka, Suita 565-0871, Japan. E-mail: inoue@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7923; Tel: +81-6-6879-7920
First published on 18th November 2015
Abstract
A chiral template was constructed from 7-ethynyl-1,5,7-trimethyl-3-azabicyclo[3.3.1]nonan-2-one by Sonogashira cross-coupling with 4,4′′-diiodoterphenyl and was shown to bind the title compound strongly by hydrogen bonding resulting in enantioselectivities of up to 55% enantiomeric excess (ee) in the [4+4] anthracene photodimerization.
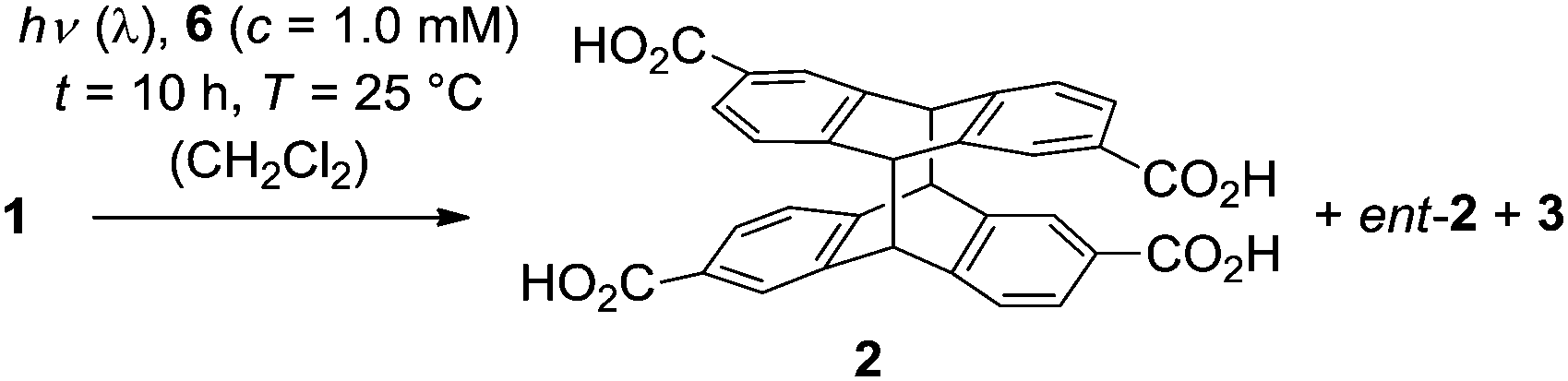
Irradiation of anthracene-2,6-dicarboxylic acid (1) in an aqueous basic solution leads to the formation of two diastereomeric [4+4] photodimerization1 products rac-2 and 3 (Scheme 1).2 The so-called anti diastereoisomer rac-2 is chiral and its two antipodes 2 and ent-23 are formed in the absence of a chiral control element in a 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, i.e. as a racemic mixture. Compound 3 is an achiral meso-compound. In contrast to the enantioselective [4+4] photodimerization of anthracene-2-carboxylic acid, which has been extensively studied in recent years,4 there have been relatively few investigations on the photodimerization of 1. In a fundamental circular dichroism (CD) study, the absolute configuration of compounds 2 and ent-2 was determined, which allows their unambiguous assignment based on their chiroptical data, e.g. the specific rotation.2,3
:1 ratio, i.e. as a racemic mixture. Compound 3 is an achiral meso-compound. In contrast to the enantioselective [4+4] photodimerization of anthracene-2-carboxylic acid, which has been extensively studied in recent years,4 there have been relatively few investigations on the photodimerization of 1. In a fundamental circular dichroism (CD) study, the absolute configuration of compounds 2 and ent-2 was determined, which allows their unambiguous assignment based on their chiroptical data, e.g. the specific rotation.2,3
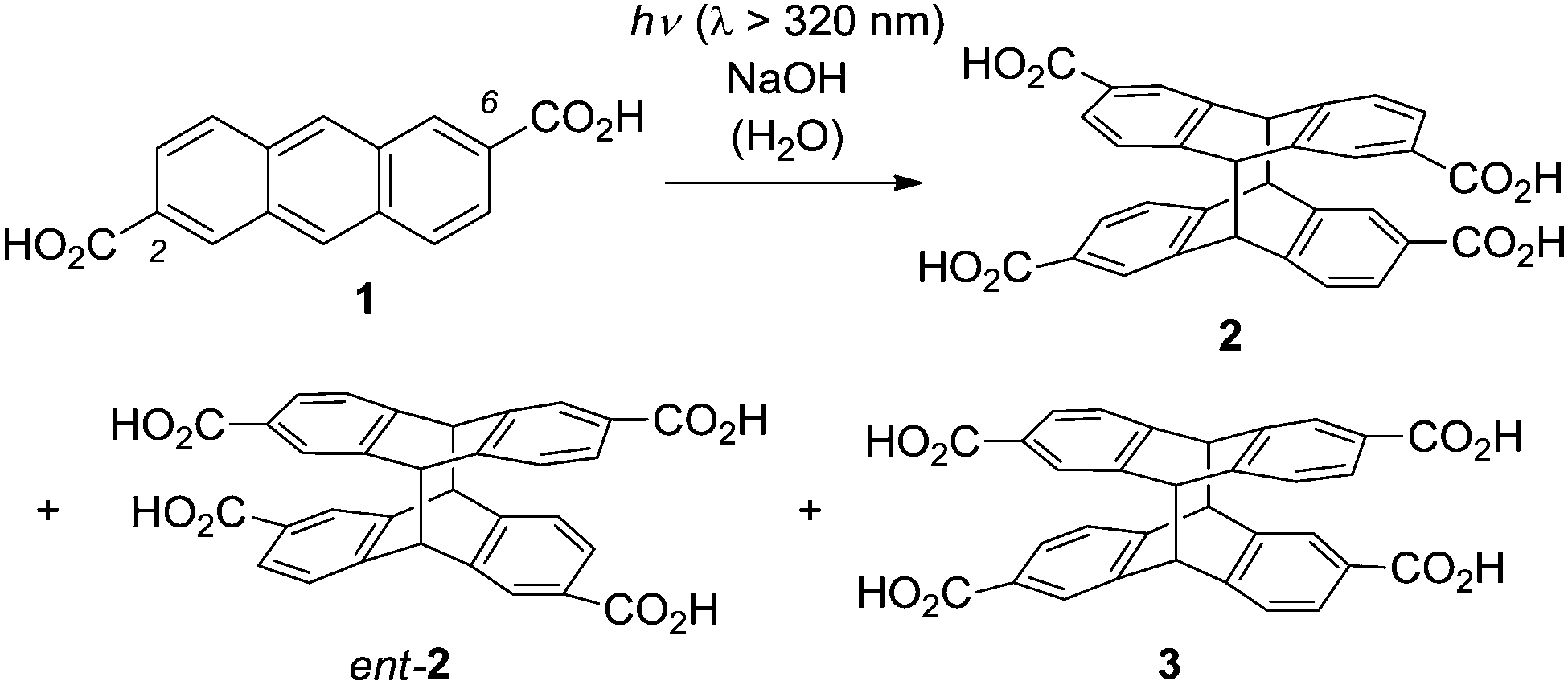 | ||
| Scheme 1 Products 2, ent-2, and 3 obtained by [4+4] photodimerization of anthracene-2,6-dicarboxylic acid (1) in an achiral environment. | ||
We became interested in the [4+4] photodimerization of diacid 1, because its two carboxylic acid groups seemed ideally positioned for a possible templation by a chiral difunctional hydrogen-bonding device. While chiral lactams with a 1,5,7-trimethyl-3-azabicyclo[3.3.1]nonan-2-one skeleton have amply shown their potential as templates for enantioselective photochemical reactions,5,6 their use is limited to nonpolar solvents such as toluene, in which the two hydrogen bonds between substrate and template are sufficiently strong7 to exert the desired directing effect. It was anticipated that dicarboxylic acid 18 would be capable of forming four hydrogen bonds to an appropriately designed chiral template with two chemically linked lactam units of identical chirality. A template of this type was readily constructed by Sonogashira cross-coupling of known 7-ethynyl-1,5,7-trimethyl-3-azabicyclo[3.3.1]nonan-2-one (4)9 with commercially available 4,4′′-diiodoterphenyl (5). Due to the low solubility of terphenyl 5 in nonpolar solvents, the use of DMF as the solvent was required for a successful transformation. In other solvents, e.g. THF, exclusive alkyne dimerization was observed (Scheme 2).
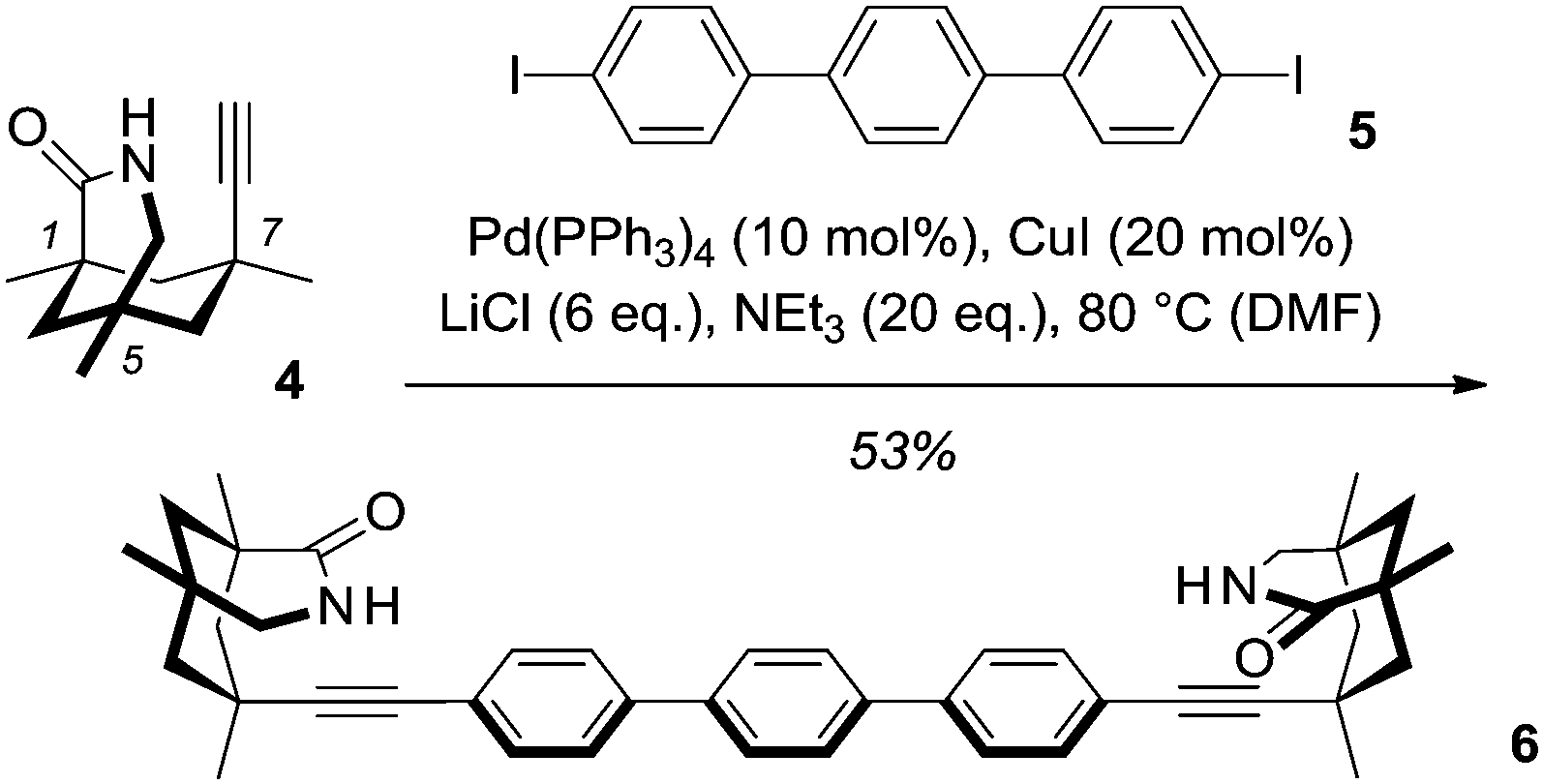 | ||
| Scheme 2 Synthesis of chelating, C2-symmetric chiral template 6 by Sonogashira cross-coupling of 4,4′′-diiodoterphenyl (5) and alkyne 4. | ||
Gratifyingly, the solubility of difunctional template 6 in organic solvents was sufficient to record its UV-Vis and fluorescence spectra. Its photophysical behaviour is dominated by the terphenyl chromophor10 and the spectra resemble the spectra of unsubstituted terphenyl (see ESI†). The singlet-state lifetime was determined by the single-photon counting method11 as 0.7 ns. In order to study the complexation between dicarboxylic acid 1 and template 6, fluorescence spectra of 1 were recorded with excitation at λexc = 388 nm (0–1′ transition of the 1Lb band12). At this wavelength, template 6 is transparent and light absorption is exclusively due to acid 1. Fluorescence spectra were measured at a fixed acid concentration of 4.95 μM in CH2Cl2/DMSO (0.02% DMSO v/v) with varying concentrations of the template. Fluorescence quenching was observed upon gradual addition of the template. Appreciable peak shifts without any isoemissive points were detected, indicating that not a single but multiple complex species are formed in the system with a weak fluorescence at longer wavelength. Least-squares-fit analysis of the intensity changes assuming a 1:1 stoichiometry enabled us to determine the apparent association constant (Ka) for complex 1·6.13 The fluorescence spectral changes at 25 °C are exemplified in Fig. 1. In an analogous fashion spectral changes were recorded at 15 °C and at 5 °C (see ESI†). As expected, the Ka values were relatively high and increased modestly by lowering the temperature; i.e. (6.6 ± 0.7) × 105 M−1 at 25 °C, (1.5 ± 0.4) × 106 M−1 at 15 °C, and (2.3 ± 0.7) × 106 M−1 at 5 °C. From the van’t Hoff plot of the temperature-dependent Ka values obtained (see ESI†), we calculated the enthalpy and entropy changes for the 1:1 complexation of 1 with 6 as ΔH° = −43 ± 8 kJ mol−1 and TΔS° = −9 ± 8 kJ mol−1 (T = 298 K). The large enthalpic gain is likely to arise from the two sets of hydrogen-bonding interactions at both ends of dicarboxylic acid 1, while the modest entropic loss is attributable to the complexation without accompanying extensive desolvation in less solvating dichloromethane.
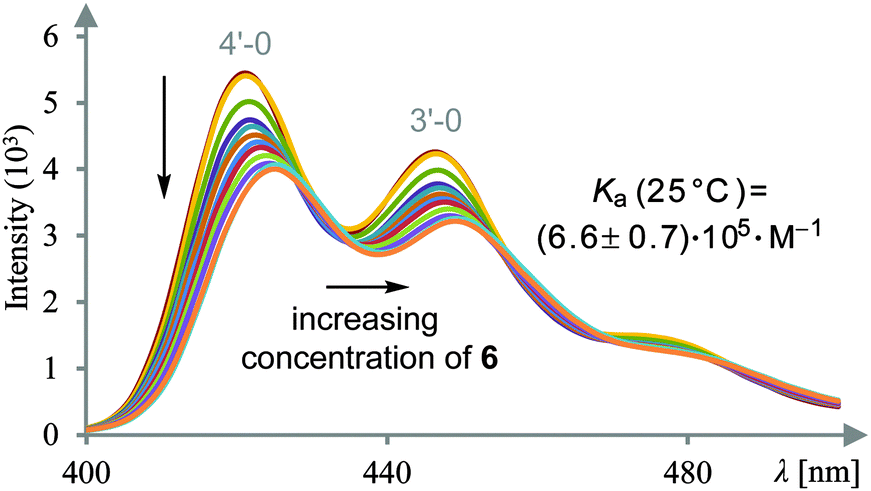 | ||
| Fig. 1 Determination of the association constant Ka for complex 1·6 at 25 °C by fluorescence spectroscopic titration at λexc = 388 nm. c (1) = 4.95 μM, c (6) = 0–24.6 μM in CH2Cl2/DMSO (0.02% DMSO v/v). | ||
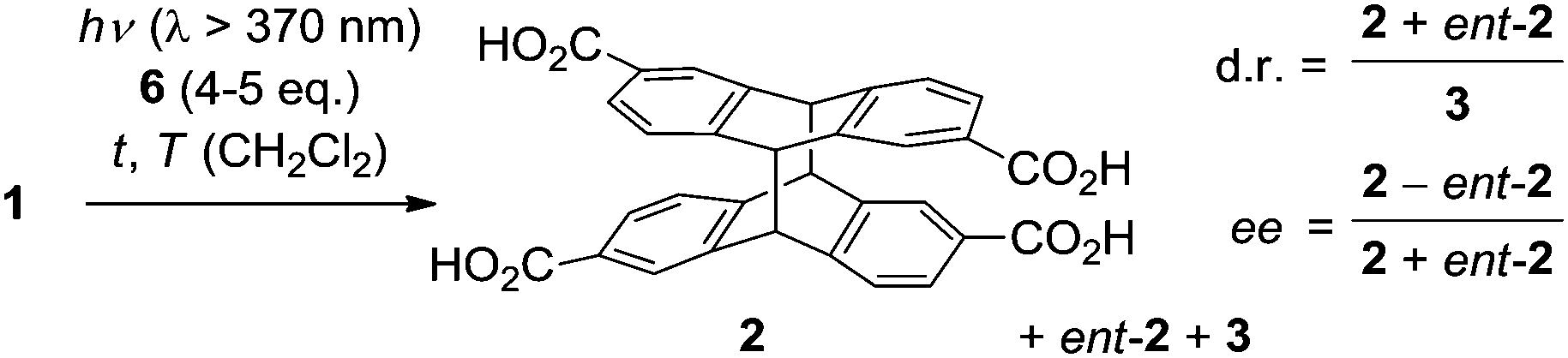
Initial studies regarding an enantioselective [4+4] photodimerization were performed at an irradiation wavelength of λ ≥ 370 nm in CH2Cl2 solution. The concentration of dicarboxylic acid 1 in solution was determined by UV-Vis spectroscopy after a given aliquot of the acid solution had been treated with a given amount of template 6, sonicated, filtered and degassed (Table 1). It was anticipated that an efficient enantioface differentiation would operate given the high association constant of complex 1·6 and given the well established high degree of steric shielding exerted by the 1,5,7-trimethyl-3-azabicyclo[3.3.1]nonan-2-one scaffold5 of the template. Unexpectedly, chiral HPLC analysis of the reaction mixture photoirradiated at 25 °C and at low substrate concentration showed the formation of essentially racemic cyclodimer 2 in addition to achiral 3 in comparable yields (entry 1). In a second experiment, the substrate concentration was increased and the reaction was performed at 0 °C (entry 2). A notable enantioselectivity was recorded and the major enantiomer was assigned to structure 2 on the basis of the known elution order for enantiomers 2 and ent-2 on a previously reported chiral HPLC phase.2 At lower temperatures, the enantioselectivity was significantly improved to give 2 in 29% enantiomeric excess (ee) at −25 °C (entry 3) and in a much higher 55% ee at −50 °C (entry 4).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | c (1) [μM] | c (6) [mM] | T [°C] | t [h] | conv.b [%] | d.rc | eec [%] |
| a The reactions were performed at the indicated temperature in quartz cuvettes employing a high pressure mercury lamp as irradiation source and a UV 37 filter (50% transmission at λ = 370 nm). b Determined by UV-Vis spectroscopy. c Determined by HPLC analysis. | |||||||
| 1 | 9.3 | 0.04 | 25 | 5 | 8 | 53/47 | <5 |
| 2 | 176 | 1.0 | 0 | 1 | 7 | 57/43 | 13 |
| 3 | 210 | 1.0 | −25 | 2 | 13 | 69/31 | 29 |
| 4 | 174 | 1.0 | −50 | 4 | 10 | 76/24 | 55 |
DFT calculations14 revealed that the coordination mode of dicarboxylic acid 1 to template 6 is more complex (Fig. 2) than originally assumed based on simple molecular models. Apparently, there are two diastereomeric conformations 1·6-I and 1·6-II, which are formally interconvertable by a rotation around the anthracene–COOH single bond.15 In addition, it was shown that also the central phenyl ring of the terphenyl unit can rotate within complexes I and II16 which eventually leads to two pairs of conformers I/I′ and II/II′ for 1·6. The energy difference between conformers I and I′ or II and II′ (Table 2) was found to be rather marginal (<0.1 kJ mol−1), while that between I and II or I′ and II′ was more significant (0.2–0.3 kJ mol−1) but still small (Table 2).
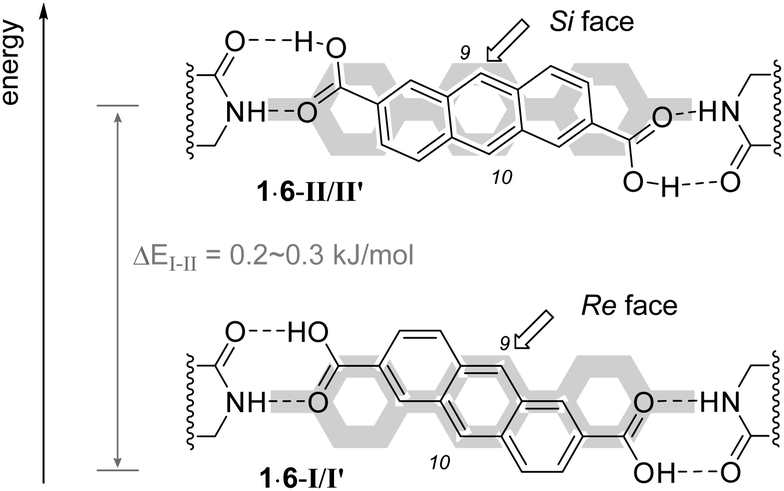 | ||
| Fig. 2 Conformations I/I′ and II/II′ for complex 1·6 and shielding by the terphenyl unit (in gray). Note that two conformations are also possible in the terphenyl unit (see narrative). | ||
| Complex | ΔE [kJ mol−1] | 1st transition (1Lb band) | 2nd transition (1La band) | ||
|---|---|---|---|---|---|
| Excitation energy [eV] | Oscillator strength | Excitation energy [eV] | Oscillator strength | ||
| a The calculations were performed at the RI-CC2/def2-TZVP//DFT-D3(BJ)-TPSS/def2-TZVP level.14,17 | |||||
| I | ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) 0 0 |
3.429 | 0.0468 | 3.712 | 0.0216 |
| I′ | 0.00 | 3.428 | 0.0468 | 3.708 | 0.0232 |
| II | +0.20 | 3.445 | 0.0497 | 3.737 | 0.0185 |
| II′ | +0.28 | 3.446 | 0.0498 | 3.739 | 0.0179 |
The fact that not a single but two pairs of conformations are located closely in energy enables facile switching of the enantiotopic face differentiation when going from I/I′ to II/II′. While the terphenyl unit shields efficiently the Si face in conformation I/I′ and attack at C9 should occur exclusively from the Re face, the opposite situation is encountered in conformation II/II′ and Si face attack should be preferred. The lower energy of conformation I/I′vs.II/II′ is in agreement with the preferred formation of enantiomer 2 in the [4+4] photodimerization at lower temperatures (Table 1), which is the result of a Re face attack. Antipodal ent-2 is formed by a Si face attack of two dicarboxylic acids, both of which adopt the Si-face exposed conformation II/II′. The achiral product 3 is likely the product of a photodimerization in which the two dicarboxylic acids are in opposing chiral complexation environments, i.e. one in conformation I/I′ and the other in conformation II/II′.
The energies of electronic transitions of the complexes were estimated at the RI-CC2/def2-TZVP level17 of theory (Table 2). Intriguingly, significant differences were found in the excitation energy of the 1Lb band (ca. 0.02 eV) between conformers I/I′ and II/II′. Although the calculated excitation energies are slightly overestimated (relative to the experiment) due to a known systematic error,2,18 it is reasonable to assume that there is a notable difference in excitation wavelength for I/I′ and II/II′ at the lowest energy band. At a wavelength of λ = 400 nm the calculated value for the excitation energy of ca. 0.02 eV corresponds to a Δλ of ca. 3 nm possibly enabling a selective excitation. To test this possibility, wavelength-selective irradiation experiments were performed employing adequate band-pass filters (Table 3). At λ = 390 nm, the enantioselectivity was expected to reflect the value previously recorded at room temperature. The fact that an ee was now notable (Table 3, entry 1) as opposed to entry 1 of Table 1 is ascribed to the higher concentration of the substrate in the latter experiment.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | c (1) [μM] | λ [nm] | conv.b [%] | d.rc | eec [%] |
| a The reactions were performed at 25 °C in quartz cuvettes employing a 300 W xenon lamp with an appropriate band-pass filter (full width at half maximum: ±10 nm) for each indicated wavelength. b Determined by UV-Vis spectroscopy. c Determined by HPLC analysis. | |||||
| 1 | 201 | 390 | 24 | 59/41 | 7 |
| 2 | 207 | 410 | 26 | 61/39 | 6 |
| 3 | 186 | 420 | 35 | 60/40 | 16 |
At λ = 410 nm (entry 2), there was a slight drop in enantioselectivity compared to λ = 390 nm (entry 1). which was, however, within the experimental error of ee determination by chiral HPLC. It was secured that there was no enantiomeric enrichment of ent-2, i.e. a negative ee. Pleasingly, irradiation at λ = 420 nm (entry 3) resulted in a significant enantioselectivity increase indicating that the enantioselectivity is wavelength dependent. In addition, due to the higher absorbance at this wavelength, the conversion of the reaction was increased compared to the entries 1 and 2. The latter results support the hypothesis, that the two conformations I/I′ and II/II′ show different UV/Vis absorption properties and that conformation I/I′ can be selectively excited.
In summary, the first enantioselective [4+4] photodimerization of anthracene-2,6-dicarboxylic acid (1) has been achieved employing a new type of chiral scaffold. Despite significant enantioselectivities for product enantiomer 2 (up to 55% ee), it was found that the fixation within the scaffold is not completely rigid but rather allows for rotation of the photochemically active entity. An intriguing collateral finding resulted from DFT calculations, which indicated that the two diastereomeric complexes I/I′ and II/II′ absorb at different wavelengths. The enantioselectivity in favor of product 2 was shown to be wavelength dependent.
This project was supported by the Deutsche Forschungsgemeinschaft (GRK 1626, Chemical Photocatalysis) and by the Alexander von Humboldt foundation (Humboldt Research Award to Y. I.).
Notes and references
- Reviews:
(a) S. M. Sieburth and N. T. Cunard, Tetrahedron, 1996, 52, 6251–6282 CrossRef CAS
; (b) S. M. Sieburth, in Synthetic Organic Photochemistry, ed. A. G. Griesbeck and J. Mattay, Dekker, New York, 2005, vol. 12, pp. 239–268 Search PubMed
- A. Wakai, H. Fukasawa, C. Yang, T. Mori and Y. Inoue, J. Am. Chem. Soc., 2012, 134, 4990–4997 CrossRef CAS PubMed
- Compound 2 has the (M)-configuration, while compound ent-2 is (P)-configured.
- Examples:
(a) Y. Kawanami, H. Tanaka, J.-i. Mizoguchi, N. Kanehisa, G. Fukuhara, M. Nishijima, T. Mori and Y. Inoue, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1411–1413 CAS
- Examples
(a) T. Bach, H. Bergmann and K. Harms, Angew. Chem., Int. Ed., 2000, 39, 2302–2304 CrossRef CAS
- Reviews on enantioselective photochemistry:
(a) Y. Inoue, Chem. Rev., 1992, 92, 741–770 CrossRef CAS
-
(a) H. Bergmann, B. Grosch, S. Sitterberg and T. Bach, J. Org. Chem., 2004, 69, 970–973 CrossRef CAS PubMed
-
(a) R. D. Broene and F. Diederich, Tetrahedron Lett., 1991, 32, 5227–5230 CrossRef CAS
-
(a) F. Voss and T. Bach, Synlett, 2010, 1493–1496 CAS
- N. Boens, W. Qin, N. Basarić, J. Hofkens, M. Ameloot, J. Pouget, J.-P. Lefèvre, B. Valeur, E. Gratton, M. vandeVen, N. D. Silva, Y. Engelborghs, K. Willaert, A. Sillen, G. Rumbles, D. Phillips, A. J. W. G. Visser, A. van Hoek, J. R. Lakowicz, H. Malak, I. Gryczynski, A. G. Szabo, D. T. Krajcarski, N. Tamai and A. Miura, Anal. Chem., 2007, 79, 2137–2149 CrossRef CAS PubMed
-
D. V. O'Connor and D. Phillips, Time-correlated single photon counting, Academic Press, London, 1984 Search PubMed
- J. R. Platt, J. Chem. Phys., 1949, 17, 484–495 CrossRef CAS
- For the determination of Ka, the fluorescence band assigned to the 4′-0 transition (4′-0) was employed. Fluorescence occurs from higher vibrational states (4′ or 3′) of S1 to the ground state of S0.
-
(a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- It remains open whether a rotation occurs within complex 1·6 or whether the equilibrium between 1·6-I and 1·6-II is established by a decomplexation-recomplexation equilibrium.
- I. Baraldi and G. Ponterini, THEOCHEM, 1985, 122, 287–298 CrossRef
-
(a) O. Christiansen, H. Koch and P. Jørgensen, Chem. Phys. Lett., 1995, 243, 409–418 CrossRef CAS
-
(a) I. Warnke and F. Furche, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 150–166 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc09107a |
| This journal is © The Royal Society of Chemistry 2016 |