
A template-free solvent-mediated synthesis of high surface area boron nitride nanosheets for aerobic oxidative desulfurization†
Peiwen
Wu‡
a,
Wenshuai
Zhu‡
*bc,
Yanhong
Chao
bc,
Jinshui
Zhang
c,
Pengfei
Zhang
c,
Huiyuan
Zhu
c,
Changfeng
Li
a,
Zhigang
Chen
b,
Huaming
Li
*d and
Sheng
Dai
*c
aSchool of Energy and Power Engineering, Jiangsu University, Zhenjiang 212013, China
bSchool of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: zhuws@ujs.edu.cn
cChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: dais@ornl.gov
dInstitute for Energy Research, Jiangsu University, Zhenjiang 212013, China. E-mail: lihm@ujs.edu.cn
First published on 16th October 2015
Abstract
Hexagonal boron nitride nanosheets (h-BNNs) with rather high specific surface area (SSA) are important two-dimensional layer-structured materials. Here, a solvent-mediated synthesis of h-BNNs revealed a template-free lattice plane control strategy that induced high SSA nanoporous structured h-BNNs with outstanding aerobic oxidative desulfurization performance.
On account of the cost-efficiency and source-abundance of oxygen, the search for a robust aerobic oxidation catalyst is a significant challenge in the catalysis community. Noble metal-based catalysts including Au,1 Pt2 and Pd3 are general catalysts toward this goal, but their high-cost seriously limits their applications. Recently, 2D graphene carbocatalysis, a low-cost and highly selective strategy, has been developed for the activation of molecular oxygen in catalysis due to its high specific surface areas (SSAs) and special layer structure.4,5 Analogously, boron nitride with a two-dimensional (2D) hexagonal structure can be a promising candidate for aerobic catalysts,6,7 but its related study is still in its infancy.
Hexagonal boron nitride nanosheets (h-BNNs) are defined as materials whose free charges are immobile in one spatial dimension, but mobile in another two. This property enables h-BNNs to have novel or superior functions, distinct from traditional bulk materials.8,9 Starting from reported applications, h-BNNs have been widely applied in catalyst support,10,11 adsorption of pollutants,12 hydrogen storage,13 drug delivery14 and water cleanup.15,16 The structural and functional properties of h-BNNs affect their performance greatly,17 especially in catalysis. Therefore, it is essential to develop new strategies for h-BNNs with high SSA, porous structures and defects.18,19
Herein, a solvent-mediated lattice plane control strategy for h-BNNs with high SSA is developed. h-BNNs are synthesized by pyrolysis of a mixture of urea and boric acid recrystallization with a solvent. In the recrystallization process, our study indicates that the low boiling point aqueous methanol solvent is the key to promote the formation of urea with high-index facets (2kl) and contributes to generating micro-/mesoporous structure h-BNNs. This nanoengineering of h-BNNs, with a high SSA of up to 1900 m2 g−1 entails a template-free pyrolysis reaction of boric acid and urea. These findings manifest that the high SSA is caused by a solvent-mediated lattice plane control mechanism. Moreover, a novel attempt to activate oxygen in the oxidation of aromatic sulfur compounds in fuel has been performed and presented outstanding catalytic performance.
To synthesize nanoporous h-BNNs, a certain molar ratio of boric acid and urea (0.01 mol of H3BO3 mixed with 0.15 mol of urea for M-BN15 and 0.30 mol of urea for other h-BNNs) in an alcohol was mixed with water (a mixed solution contains 20 mL of alcohols and 20 mL of water) for recrystallization. A white crystalline powder formed by evaporating solvents was pyrolyzed under the protection of N2 to produce h-BNNs. The sample notation, detailed synthesis conditions and BET surface area for the nanoporous h-BNNs are listed in Table 1.
| Samples | Solvents (volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
Molar ratio (boric acid:urea) |
BET surface area (m2 g−1) |
|---|---|---|---|
| BN-M15 | Methanol/water | 1:15 |
1290 |
| BN-M30 | Methanol/water | 1:30 |
1900 |
| BN-E30 | Ethanol/water | 1:30 |
641 |
| BN-P30 | Propanol/water | 1:30 |
539 |
| BN-B30 | Butanol/water | 1:30 |
239 |
| BN-W30 | Water | 1:30 |
483 |
The as-prepared h-BNNs were examined by N2 adsorption–desorption and corresponding Barrett–Joyner–Halenda (BJH) pore-size distributions. All isotherms in Fig. 1a belong to type II based on the IUPAC classification, and type H3 hysteresis loops in the relative pressure range of 0.4–1.0 indicate the presence of mesopores with a diameter of about 2 nm (Fig. 1b). Notably, in the low relative pressure region, the isotherms rise sharply, indicating the presence of microporous structures. Moreover, our methanol-mediated h-BNNs highlight the prominent formation of highly microporous structures, while a significantly lower microporosity was observed for the h-BNNs synthesized with other aqueous alcohols or water as solvents. Correspondingly, the SSAs of BN-M15 and BN-M30 are as high as 1290 and 1900 m2 g−1, respectively, while those of the nanoporous h-BNNs surprisingly decrease with the increasing boiling point of alcohols (Fig. 1c), in the range of 239–641 m2 g−1. These results indicate that the solvent boiling point is inversely correlated with the SSAs of the resulting h-BNN products.
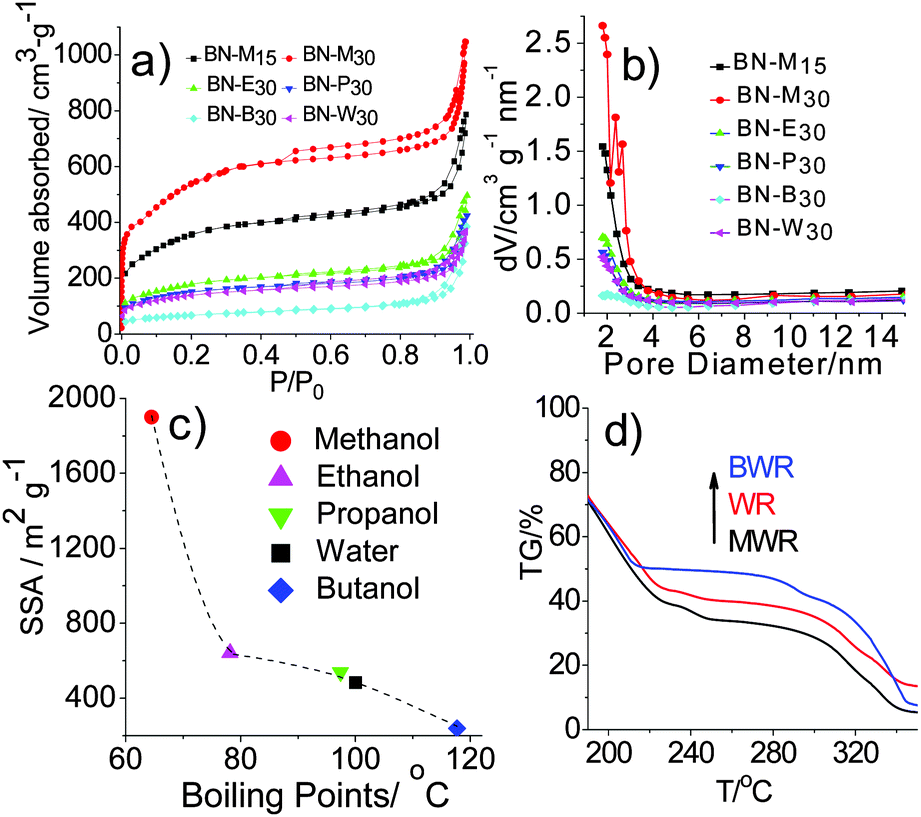 | ||
| Fig. 1 (a) N2 adsorption–desorption isotherm of h-BNNs; (b) corresponding BJH pore-size distributions of h-BNNs; (c) relationship of solvent boiling points with BET of h-BNNs; (d) TG analysis of precursors of h-BNNs (BWR, WR and MWR). | ||
Why do solvents with different boiling points cause such a large difference in the SSAs of nanoporous h-BNNs? In order to make sense of this problem, we choose three representative precursor formulations corresponding to the highest, middle-bound and lowest values of h-BNN SSAs. They are boric acid and urea with a molar ratio of 1:30 dissolved in methanol/water (MW), pure water (W) and butanol/water (BW). Subsequently we evaporate solvents for recrystallization, producing white solids, denoted as MWR, WR and BWR, respectively. Their chemical structures are characterized by both FT-IR and Raman spectra (Fig. S1, ESI†), showing no obvious differences. The results indicate that solvents have no effects on the chemical compositions of the precursors. However, TG analysis of three precursors shows that their decomposition rates are different after 200 °C (Fig. 1d). Thus, their structures are characterized by XRD patterns (Fig. 2), which are indexed to the standard PDF card for urea (PDF#83-1436). The disappearance of H3BO3 peaks is assigned to its low content. The peaks at 22.3°, 24.6°, 29.3°, 31.7° and 35.5° are attributed to the (110), (101), (111), (200) and (210) lattice planes of urea, respectively. Notably, the intensity (I(hkl)) ratios of different facets in MWR, WR and BWR are listed in Fig. 2 and the results show that the relative intensities of the low-index facet (110) and (101) peak weaken gradually in the order of MWR, WR and BWR, while the high-index facets (200) and (210) are enhanced in the same order. In XRD patterns, relative intensities reflect preferred lattice plane orientation. Interestingly, the urea precursor with high-index facets shows a negative correlation with the boiling point of the solvents. A lower boiling point of a solvent can produce relatively higher intensities of the high-index facets. As we know, the crystal growth rate highly relies on the evaporation rate of solvents (depending on boiling points) and is of great importance in control of crystal orientations. The introduction of a low boiling-point solvent can accelerate the recrystallization rate and further tame the recrystallization of urea along a preferred orientation of the (2kl) lattice plane, leading to urea with greater intensity of high-index facets. Combined with the N2 adsorption–desorption isotherm, it is evident that the introduction of low-boiling-point aqueous methanol is conducive to formation of urea with high-index facets, further triggering the formation of higher SSA h-BNNs.
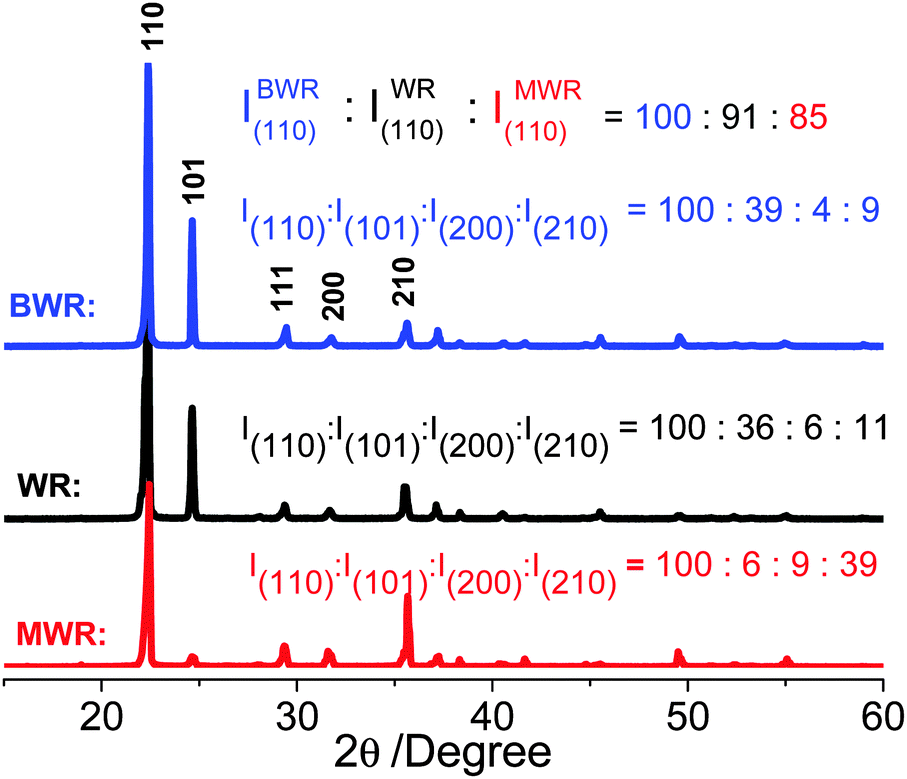 | ||
| Fig. 2 XRD patterns of BWR, WR and MWR precursors. | ||
The rationales behind this solvent-mediated formation of high SSA h-BNNs with a controllable lattice plane are two fold: (i) construction of a few-layer structure and (ii) an abundant nanoporous structure. Based on crystallography, higher-index facets possess higher crystal surface energies.20,21 Thus, the urea with high-index facets is more active, which is more likely to produce reactive intermediates (cyanuric acid). TG analysis (Fig. 1d) of the pyrolysis rate of precursors follows the order of MWR > WR > BWR, which is a supporting evidence for the above results. Moreover, higher-index facets mean smaller atom density and larger distances between two atoms. Urea molecules in higher-index facets are thus further apart from each other. When the urea is polymerized, smaller atom density intermediates are formed on the higher-index surface, which is in favor of the formation of fewer-layer h-BNNs and a larger interlayer distance in pyrolysis, which is also proved by XRD patterns (Fig. S2a, ESI†). The peak of (002) in XRD shows a slight shift to low angle when high-index facets are enhanced in the urea, indicating that the layer–layer distance of h-BNNs is extended.15
The morphologies and fine structures of the as-prepared h-BNNs are also carefully determined by field emission scanning electron microscopy (SEM) and high resolution transmission electron microscopy (HRTEM), showing that the h-BNNs represent a 2D structure with wrinkles (Fig. 3a and b and Fig. S3, ESI†). Meanwhile, it is clearly observed that all samples contain curly and flat layers, and the number of layers increases with the increasing solvent boiling points. Moreover, HRTEM of BN-M30 in Fig. 3c shows two or three parallel fringes, indicating that the numbers of layers of BN-M30 are mostly 2 and 3. In addition, there exist some arches and amorphous areas (Fig. 3c), illustrating the existence of defects.22 Furthermore, atomic resolution scanning transmission electron microscopy (STEM) is performed (Fig. 3d) in which the resolved features correspond to the hexagonal lattice. Meanwhile, vacancies can be seen in Fig. 3d (red circles), consistent with the results shown in Fig. 3c. The fast Fourier transform (FFT) analysis in Fig. 3e shows three sets of hexagonal spots, which also indicate that the region of Fig. 3e has few-layers.23 Besides, the distance between the two neighbouring bright reflections in the inset of Fig. 3e perfectly agrees with the B–B or N–N atom separations in h-BN,24 further confirmed by the X-ray diffraction (XRD) profile of the (100) lattice (Fig. S2a, ESI†). The atomic force microscopy (AFM) image further represents that the height of BN-M30 is about 2 nm (Fig. 3f). In AFM images, the thickness of monolayer BN is about 1 nm due to trapped solvents between the layers and the underlying substrate,25 which is in good agreement with HRTEM. Besides the lattice plane control mechanism, BN-M30 has a larger SSA than BN-M15 because fewer-layer BNNs were produced via increasing the proportion of urea in the reaction mixture.26
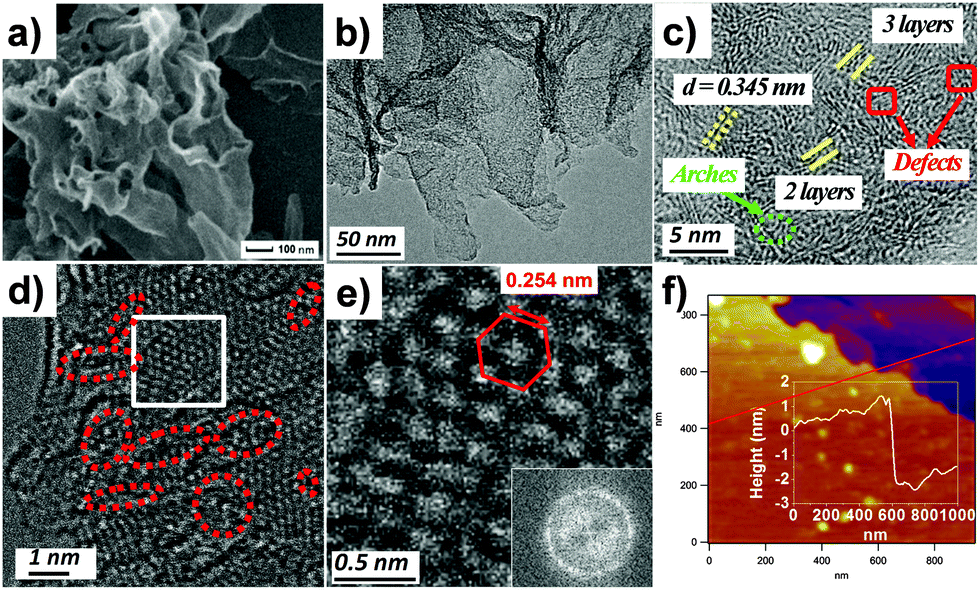 | ||
| Fig. 3 Morphologies of BN-M30. (a) SEM image; (b and c) HRTEM images; (d) STEM image of M-BN30; (e) magnified STEM image of the white area in (d) and the inset in (e) shows the FFT pattern of panel (e); (f) AFM image and corresponding height profile. | ||
Additionally, in the case of point (ii), abundant nanoporous structures also play a vital role in the high SSA. Combined with the intricate urea pyrolysis process,27 we proposed that the formation procedure of h-BNNs included six main stages (Fig. S4, ESI†). When the precursor was heated to ∼200 °C, urea polycondensed to form cyanuric acid (C3H3N3O3) and then reacted with B2O3 to produce h-BNNs.28 Proverbially, high-index lattice planes possess high crystal surface energies. Urea with a high-index lattice plane is more active, which may react with B2O3 preferentially and generate h-BNNs. When the temperature was increased unceasingly, urea with a low crystal surface energy may polycondense sequentially and produces numerous gases such as NH3, CO2 and H2O to fabricate micro/mesopores in the already-formed h-BNNs. This proposal was proved by thermogravimetric (TG) analysis (Fig. 1d). In TG analysis, the decomposition step occurs at around 200 °C, which may be ascribed to the polycondensation of urea with high-index lattice planes and show decreasing mass loss obviously in MWR, WR and BWR. A detailed formation process is presented in Scheme 1. These findings are also in accordance with the BJH pore-size distributions of samples (Fig. 1b).
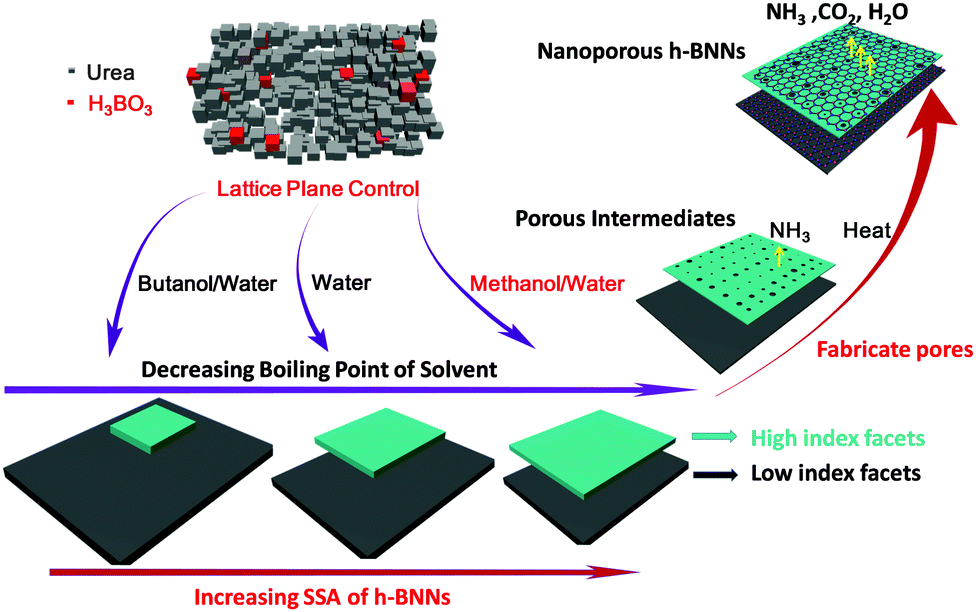 | ||
| Scheme 1 Solvent-mediated lattice plane control process for high SSA nanoporous h-BNNs. | ||
To further investigate the effect of the h-BNNs structure and composition on their SSAs, X-ray photoelectron spectroscopy (XPS) (Fig. S5–S7 and Table S1, ESI†) demonstrates that chemical states of all h-BNNs show no obvious differences, indicating that the compositions of h-BNNs are not the origin of the difference observed in SSAs, which is further proved by spectral characterization including XRD patterns, Raman spectra, FT-IR spectra and UV-Vis DRS spectra in Fig. S2 (ESI†).
In the past few decades, regulations on sulfur compounds in fuel oil have become increasingly stringent in many countries.29,30 The aerobic oxidative desulfurization of aromatic sulfur compounds with a metal-free catalyst is a transformative way but a formidable challenge.31–33 Herein, high SSA porous h-BNNs were used as metal-free catalysts for aerobic oxidative deep-desulfurization and showed efficient catalytic performance with dibenzothiophene (DBT) removal of 84.8–98.8% (Fig. 4a). BN-M30 exhibited the best catalytic activity of 98.8% for oxidizing DBT to DBTO2 (Fig. S8, ESI†), assigned to the highest SSA. The defects in the h-BNNs may also contribute to the high catalytic activity.34 Compared with the harsh reaction conditions (∼350 °C, ∼10 MPa) of hydrogen desulfurization, this aerobic oxidation is safe and cost-efficient. Additionally, this BN-M30 metal-free catalyst can be recycled 10 times with sulfur removal more than 90% (Fig. S9, ESI†), indicating its promising application in aerobic catalysis.
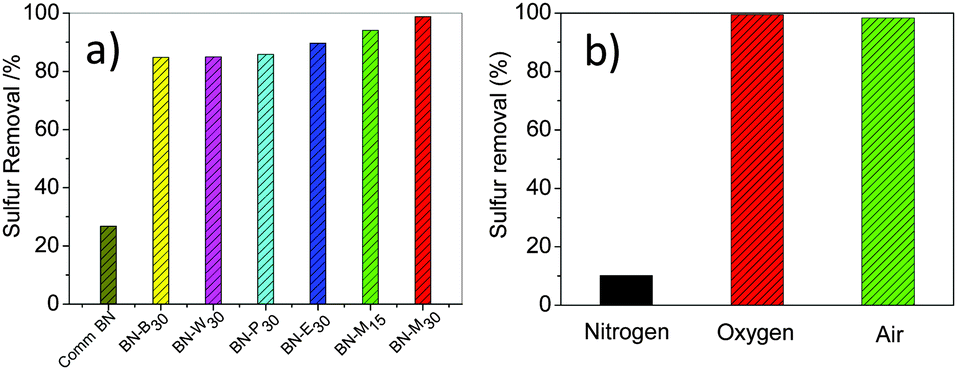 | ||
| Fig. 4 (a) Catalytic activities of h-BNNs for the aerobic oxidation of DBT; (b) effect of oxidants on the aerobic oxidation of DBT. For reaction conditions see the Experimental section in the ESI.† | ||
To further verify the above aerobic catalysis, comparative experiments with commercially available BN (Comm-BN) and different oxidants were carried out. Less than 30% of DBT was removed catalysed by Comm-BN for aerobic oxidative desulfurization, far away from the prepared h-BNNs. Structurally, the Comm-BN is highly crystalline without defects and its BET is only ∼10 m2 g−1, while the porous h-BNNs in this work exhibit high SSA and abundant structural defects. Moreover, upon bubbling N2 (Fig. 4b), the sulfur removal is only 10.2% with BN-M30 as a catalyst, which may be ascribed to the adsorption capability of BN-M30. Additionally, under the same reaction conditions, oxygen or air was used instead of nitrogen, reaching 99.4% or 98.8% sulfur removal, respectively. These results demonstrate that h-BNNs react with oxygen in air to generate active intermediates, which subsequently oxidize DBT.
In summary, we have successfully synthesized h-BNNs with rather high SSA (1900 m2 g−1) through a template-free solvent-mediated nanoengineering strategy. A novel lattice plane control mechanism was proposed to illustrate the formation process. Moreover, the h-BNNs presented high catalytic activity in aerobic oxidative deep-desulfurization. This work not only sheds new light on the structural control of h-BNNs through the judicious choice of precursor structures, but also triggers a new strategy for the large-scale preparation of high SSA nanomaterials.
We thank the National Nature Science Foundation of China (No. 21376111, 21276117 and 21576122), Six Big Talent Peak in Jiangsu province (JNHB-004), and the Postgraduate Innovation Project of Jiangsu Province (No. KYLX15_1067). SD was sponsored by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, U.S. Department of Energy.
Notes and references
- A. Corma, P. Concepcion, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadas, M. A. Lopez-Quintela, D. Buceta, E. Mendoza, G. Guilera and A. Mayoral, Nat. Chem., 2013, 5, 775–781 CrossRef CAS PubMed.
- H. Yuan, W. J. Yoo, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2012, 134, 13970–13973 CrossRef CAS PubMed.
- Z.-A. Qiao, P. Zhang, S.-H. Chai, M. Chi, G. M. Veith, N. C. Gallego, M. Kidder and S. Dai, J. Am. Chem. Soc., 2014, 136, 11260–11263 CrossRef CAS PubMed.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- J. L. Long, X. Q. Xie, J. Xu, Q. Gu, L. M. Chen and X. X. Wang, ACS Catal., 2012, 2, 622–631 CrossRef CAS.
- K. Uosaki, G. Elumalai, H. Noguchi, T. Masuda, A. Lyalin, A. Nakayama and T. Taketsugu, J. Am. Chem. Soc., 2014, 136, 6542–6545 CrossRef CAS PubMed.
- W. W. Lei, D. Portehault, R. Dimova and M. Antoniettit, J. Am. Chem. Soc., 2011, 133, 7121–7127 CrossRef CAS PubMed.
- A. Pakdel, Y. Bando and D. Golberg, Chem. Soc. Rev., 2014, 43, 934–959 RSC.
- Y. Lin and J. W. Connell, Nanoscale, 2012, 4, 6908–6939 RSC.
- W. S. Zhu, B. L. Dai, P. W. Wu, Y. H. Chao, J. Xiong, S. H. Xun, H. P. Li and H. M. Li, ACS Sustainable Chem. Eng., 2015, 3, 186–194 CrossRef CAS.
- C. J. Huang, W. Q. Ye, Q. W. Liu and X. Q. Qiu, ACS Appl. Mater. Interfaces, 2014, 6, 14469–14476 CAS.
- J. Xiong, W. S. Zhu, H. P. Li, W. J. Ding, Y. H. Chao, P. W. Wu, S. H. Xun, M. Zhang and H. M. Li, Green Chem., 2015, 17, 1647–1656 RSC.
- Q. H. Weng, X. B. Wang, Y. Bando and D. Golberg, Adv. Energy Mater., 2014, 4 DOI:10.1002/aenm.201301525.
- Q. H. Weng, B. J. Wang, X. B. Wang, N. Hanagata, X. Li, D. Q. Liu, X. Wang, X. F. Jiang, Y. Bando and D. Golberg, ACS Nano, 2014, 8, 6123–6130 CrossRef CAS PubMed.
- W. W. Lei, D. Portehault, D. Liu, S. Qin and Y. Chen, Nat. Commun., 2013, 4, 1777 CrossRef PubMed.
- Y. L. Yu, H. Chen, Y. Liu, V. S. J. Craig, C. M. Wang, L. H. Li and Y. Chen, Adv. Mater. Interfaces, 2015, 2 DOI:10.1002/admi.201400529.
- M. Maleki, A. Beitollahi and M. Shokouhimehr, Eur. J. Inorg. Chem., 2015, 2478–2485 CrossRef CAS.
- A. Dhakshinamoorthy, A. Primo, I. Esteve-Adell, M. Alvaro and H. Garcia, ChemCatChem, 2015, 7, 776–780 CrossRef CAS.
- Y. H. Zhang, X. F. Weng, H. Li, H. B. Li, M. M. Wei, J. P. Xiao, Z. Liu, M. S. Chen, Q. Fu and X. H. Bao, Nano Lett., 2015, 15, 3616–3623 CrossRef CAS PubMed.
- N. F. Yu, N. Tian, Z. Y. Zhou, L. Huang, J. Xiao, Y.-H. Wen and S.-G. Sun, Angew. Chem., Int. Ed., 2014, 53, 5097–5101 CAS.
- H. B. Zhang, Y. G. Lu, H. Liu and J. Z. Fang, Nanoscale, 2015, 7, 11591–11601 RSC.
- R. H. Que, Y. C. Huang, Q. L. Li, H. Yao, B. Y. Geng and M. W. Shao, ACS Appl. Mater. Interfaces, 2014, 6, 19752–19757 CAS.
- L. Song, L. J. Ci, H. Lu, P. B. Sorokin, C. H. Jin, J. Ni, A. G. Kvashnin, D. G. Kvashnin, J. Lou, B. I. Yakobson and P. M. Ajayan, Nano Lett., 2010, 10, 3209–3215 CrossRef CAS PubMed.
- C. Y. Zhi, Y. Bando, C. C. Tang, H. Kuwahara and D. Golberg, Adv. Mater., 2009, 21, 2889–2893 CrossRef CAS.
- M. Du, X. Li, A. Wang, Y. Wu, X. Hao and M. Zhao, Angew. Chem., Int. Ed., 2014, 53, 3645–3649 CrossRef CAS PubMed.
- A. Nag, K. Raidongia, K. P. S. S. Hembram, R. Datta, U. V. Waghmare and C. N. R. Rao, ACS Nano, 2010, 4, 1539–1544 CrossRef CAS PubMed.
- P. A. Schaber, J. Colson, S. Higgins, D. Thielen, B. Anspach and J. Brauer, Thermochim. Acta, 2004, 424, 131–142 CrossRef CAS.
- P. W. Wu, W. S. Zhu, A. M. Wei, B. L. Dai, Y. H. Chao, C. F. Li, H. M. Li and S. Dai, Chem. – Eur. J., 2015, 21, 15421–15427 CrossRef CAS PubMed.
- H. Y. Lv, P. Li, C. Deng, W. Ren, S. Wang, P. Liu and H. Zhang, Chem. Commun., 2015, 51, 10703–10706 RSC.
- W. Zhu, C. Wang, H. Li, P. Wu, S. Xun, W. Jiang, Z. Chen, Z. Zhao and H. Li, Green Chem., 2015, 17, 2464–2472 RSC.
- J. Xiao, S. Sitamraju, Y. S. Chen, S. Watanabe, M. Fujii, M. Janik and C. S. Song, AIChE J., 2015, 61, 631–639 CrossRef CAS.
- H. Y. Lv, W. Z. Ren, W. P. Liao, W. Chen, Y. Li and Z. H. Suo, Appl. Catal., B, 2013, 138, 79–83 Search PubMed.
- W. Zhang, H. Zhang, J. Xiao, Z. X. Zhao, M. Yu and Z. Li, Green Chem., 2014, 16, 211–220 RSC.
- J. X. Zhao, H. X. Wang, Y. J. Liu, Q. H. Cai and X. Z. Wang, RSC Adv., 2013, 3, 4917–4926 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc07830j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |