
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe regioselective iodination of quinolines, quinolones, pyridones, pyridines and uracil†
Uttam
Dutta
abc,
Arghya
Deb
a,
David W.
Lupton
*bc and
Debabrata
Maiti
*ab
aDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai - 400076, India. E-mail: dmaiti@chem.iitb.ac.in
bIITB-Monash Research Academy, CSE Building, IIT Bombay, Powai, Mumbai - 400076, India
cSchool of Chemistry, Monash University, Clayton 3800, Victoria, Australia. E-mail: david.lupton@monash.edu
First published on 16th October 2015
Abstract
A radical based direct C–H iodination protocol for quinolines, quinolones, pyridones, pyridines, and uracil has been developed. The iodination occurs in a C3 selective manner for quinolines and quinolones. Pyridones and pyridines undergo C3 and C5 iodination, while dimethyl uracil undergoes C5 iodination. Scope of the method was demonstrated through the rapid synthesis of both electron rich as well as electron poor heteroaromatic iodides. The protocol was found to be scalable and general, while a mechanism has been proposed.
Iodine containing compounds are integral to synthetic organic chemistry. Beyond applications in traditional synthetic methods such as metalation1 and aromatic nucleophilic substitution,2 they are now ubiquitous in cross-coupling chemistry.3 Furthermore, radiolabeled iodide analogues play a vital role in medicinal and radiotherapeutic science.4 As a consequence, extensive efforts have been made to develop useful protocols for preparing aryl iodides.5–7 However, their synthesis remains difficult, with limitations relating to the use of expensive transition metals, need for highly polar solvents, prefunctionalization, and modest regioselectivity, plaguing many reported approaches.8 On contrary to simple aryl iodide, the synthesis of heteroaromatic iodides is increasingly difficult.
Quinoline, pyridone and other nitrogen containing heterocyclic iodides are highly important structural motifs due to their presence in innumerable natural products and pharmaceutical agents.9 In recent years, much efforts have been devoted towards the regioselective synthesis of iodinated heterocycles.10–12 Aromatic Finkelstein reaction from bromides (i.e. 1) have developed as popular method for accessing iodo-quinolines (2) (eqn (1), Scheme 1).13 Recently Li et al. have disclosed a photo induced metal free Finkelstein reaction to access C3 and C4 iodinated quinoline.14 Unfortunately, such an approach demands prefunctionalization, and therefore limit generality. Direct regioselective functionalization of heterocycles arguably has the greatest potential to deliver broadly applicable iodination methods. However, selectivity with substrates bearing multiple C–H bonds makes this approach more challenging.
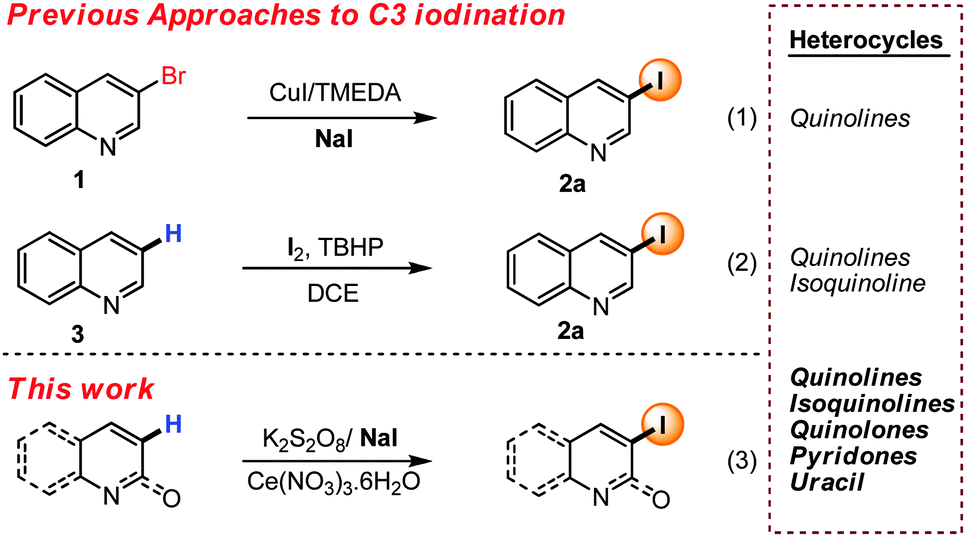 | ||
| Scheme 1 C3 selective iodination by C–H functionalization. | ||
With pyridyl and quinoline, halogenation can be addressed by exploiting the N-oxide thereby allowing selective C2 halogenation.15 While in an orthogonal approach recently reported by Chang and co-workers C8 iodination was achievable by rhodium catalyzed C–H iodination of quinoline-N-oxide with NIS.16 While selective C2 and C8 halogenation of quinolines can be achieved, mild regioselective methods for other iodinations remain limited. As part of broader studies on the functionalization of heterocycles17 in 2013, we commenced studies focused on C3 selective halogenation of quinolines and related heterocycles. We envisaged a direct radical iodination approach enabled by the mild generation of the iodo radical. It was postulated that such an approach should allow predictable C3 iodination due to the stability of the first formed radical intermediate. Very recently a related concept was communicated by Sun and Jain which allowed the iodination of different quinoline derivatives in a regioselective manner (3) (eqn (2), Scheme 1).10f,g Stimulated by this report we wish to report our approaches on this topic. While our conditions are related to those of Sun and Jain, we have been able to achieve mild iodination of both electron rich and poor quinolines, as well as pyridones, uracil and pyridines. Mechanistically we believe that in situ generation of the iodo radical leads to selective C3 iodination, although with highly electron rich substrates alternate mechanistic pathways are possible.
To achieve selective C3 iodination we commenced by reacting quinoline with K2S2O8 and sodium iodide in the presence of MnSO4 in dichloroethane (DCE) heated at 130 °C. Unfortunately, these conditions, and those in which the manganese was replaced by either tin or cobalt failed to provide 3-iodo-quinoline (2a) (Table 1, entries 1–3). In contrast the reaction in the presence of either bismuth, nickel or cerium salts gave promising yields of the expected product with Ce(NO3)3.6H2O optimal with 40% yield of 2a (Table 1, entries 4–6). Addition of 1 equivalent of TFA increased the yield, while an examination of alternate oxidants confirmed that potassium peroxodisulphate was ideal (Table 1, entry 7), although other common oxidants were also viable (Table 1, entries 8–11). Finally, increasing the stoichiometry of sodium iodide to three equivalents increased the yield further to 85% (Table 1, entry 12). Finally, control experiments demonstrated that each reagent is necessary for the formation of 3-iodoquinoline in synthetically useful yields (See ESI† for detailed optimization).
|
|
||||
|---|---|---|---|---|
| Entry | Metal salt | Oxidant | Solventa | Yieldb |
| a All reactions performed at 130 °C. b GC yield except as noted. c 1 equiv. TFA added. d 3 equiv. NaI. e Isolated yield. | ||||
| 1 | MnSO4·H2O | K2S2O8 | DCE | — |
| 2 | SnCl2·2H2O | K2S2O8 | DCE | — |
| 3 | CoCl2·6H2O | K2S2O8 | DCE | 1 |
| 4 | Bi(NO3)3·5H2O | K2S2O8 | DCE | 30 |
| 5 | Ni(NO3)2 | K2S2O8 | DCE | 37 |
| 6 | Ce(NO3)3·6H2O | K2S2O8 | DCE | 40 |
| 7c | Ce(NO3)3·6H2O | K2S2O8 | DCE | 62 |
| 8c | Ce(NO3)3·6H2O | TBHP | DCE | 61 |
| 9c | Ce(NO3)3·6H2O | K2S2O8 | 1,2,3-TCP | 45 |
| 10c | Ce(NO3)3·6H2O | K2S2O8 | t BuOH | 20 |
| 11c | Ce(NO3)3·6H2O | DTBP | DCE | 59 |
| 12 , | Ce(NO 3 )3·6H2O | K 2 S 2 O 8 | DCE | 85(78) |
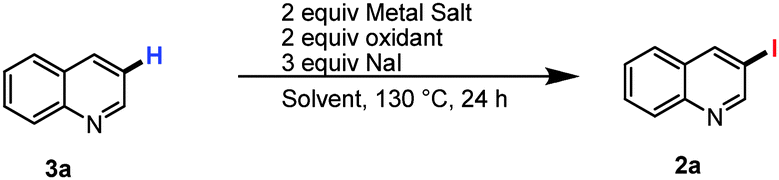
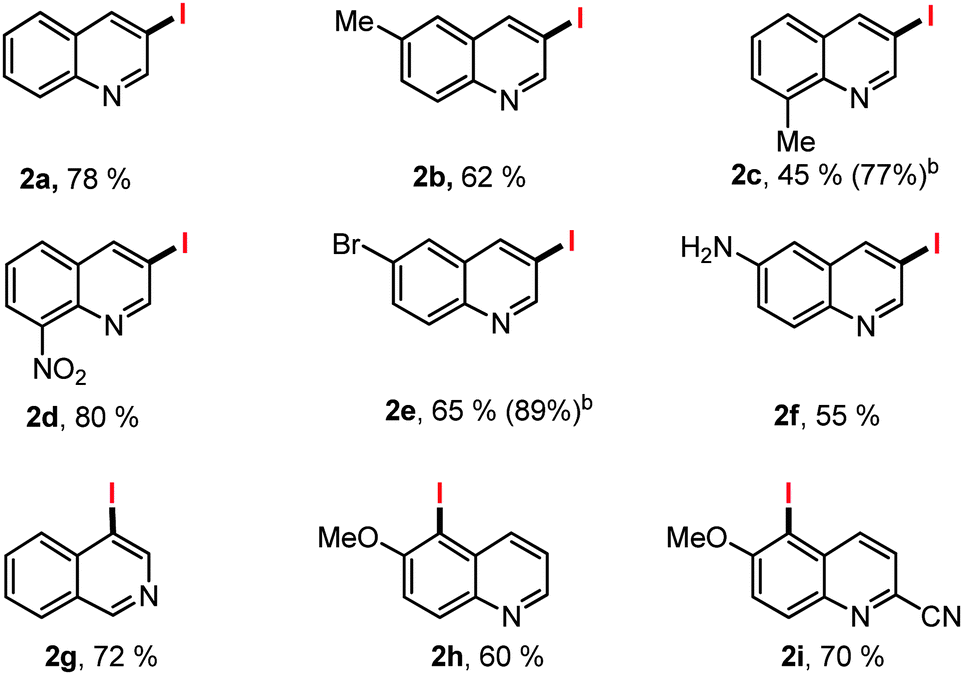
With the optimized reaction condition, the generality of the regioselective iodination was examined with the halogenation of various quinoline derivatives (Table 2). In contrast to the studies of Sun and Li who reported solely the iodination of electron poor substrates our conditions allowed 6-methyl (2b, 62%) and 8-methyl (2c, 45%), as well as electron deficient 8-NO2 (2d, 80%) iodoquinolines to be prepared in acceptable isolated yields after column chromatography. Similarly 6-bromoquinoline and electron rich 6-amino quinoline gave C3 iodinated 2e and 2f in 65% and 55% yields respectively. Isoquinoline gave C4-iodinated product 2g in 72% yield, as reported by Sun and Li. When the 6-methoxy quinolines were examined the selectivity switches to C5-iodinated products (2h, 60% and 2i, 70%).
Next, we thought to examine the related iodination of N-benzyl quinolones (i.e.4a) under the optimized reaction condition (Table 3). To our delight, we obtained the C3 iodinated N-benzyl quinolone derivative 5a in 65% yield.
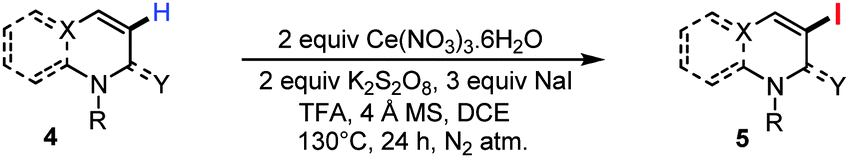
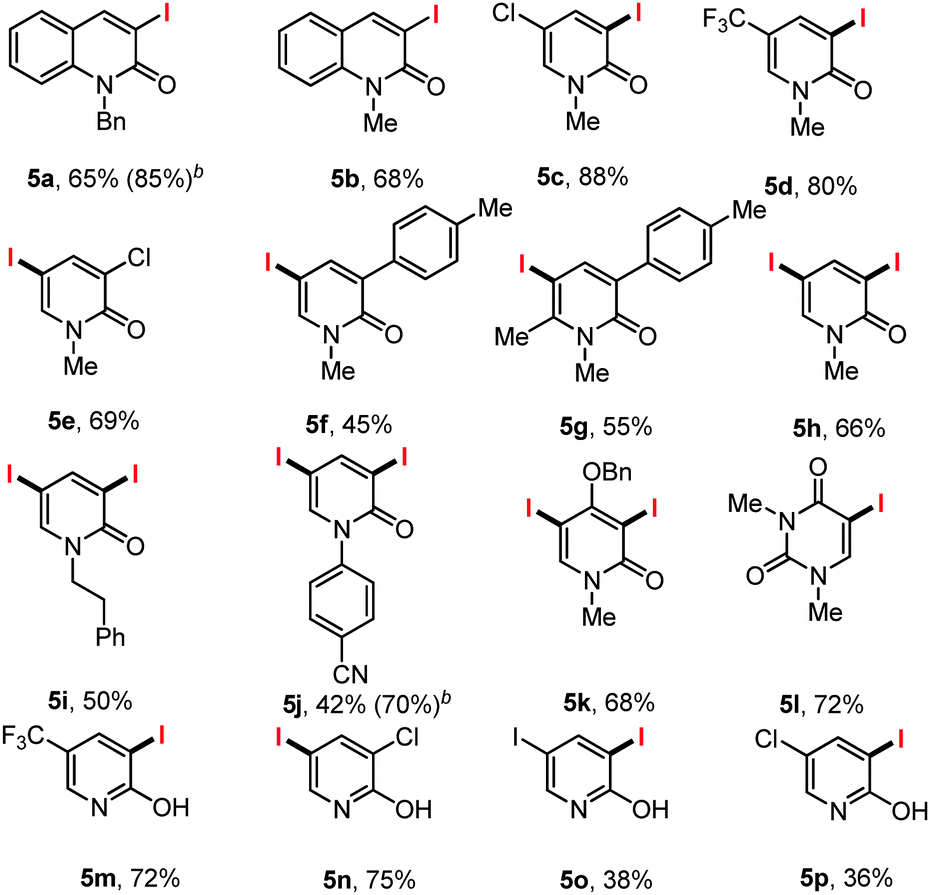
Similar results were obtained in the preparation of the N-methyl quinolone 5b, while C5 blocked (Cl, 4c or CF3, 4d) iodinated pyridones (5c and 5d) were also prepared in excellent yield.
When C3 blocked pyridones were subjected to the reaction condition (Cl, 4e; p-tolyl, 4f) the C5 iodinated products formed smoothly. The reaction was insensitive to steric congestion with the C5 iodinated C6-methyl-C3-p-tolyl pyridone prepared in 55% yield.
With N-phenylethyl, N-methyl or N-aryl pyridones bearing no additional substituents the diiodinated pyridones 5h, 5i and 5j were prepared in 66, 50 and 42% yields respectively. The electron deficient pyridone 4j was diiodinated to give 5j smoothly, while dimethyl uracil (4k) provided the product of monoiodination. Finally a series of 2-hydroxy iodopyridines 5m–5p bearing either C3 or C5 substituents were smoothly formed in acceptable yields.
To assess the scalability of the protocol conversion of 8-NO2 quinoline to 3-iodo-8-nitro quinoline (2d) was performed with 1.3 g of substrate. The expected iodide 2d was obtained in 77% yield, only slightly lower than the yield achieved with the submillimolar scale reaction (Scheme 2).
 | ||
| Scheme 2 Scaled up iodination of 8-nitro quinoline. | ||
In order to gain insight into the reaction mechanism, a number of control experiments were performed. When a radical quencher (e.g. TEMPO) was introduced this suppressed the iodination reaction with only a trace of the iodinated product of quinoline 2a. Based on this observation, it may be assumed that one of the steps for iodination is proceeding via a radical pathway.
Previous studies with pyridone demonstrated selective functionalization at C3 position via a radical based transformation.18 The observed C5-functionalized products, as in 6-methoxy quinoline 2h, likely forms by electrophilic iodination. Therefore, the present protocol can promote iodination reaction both by a radical or electrophilic path. By considering the C3 and C5 selectivity for quinoline and pyridone, a mechanism is proposed in Scheme 3.
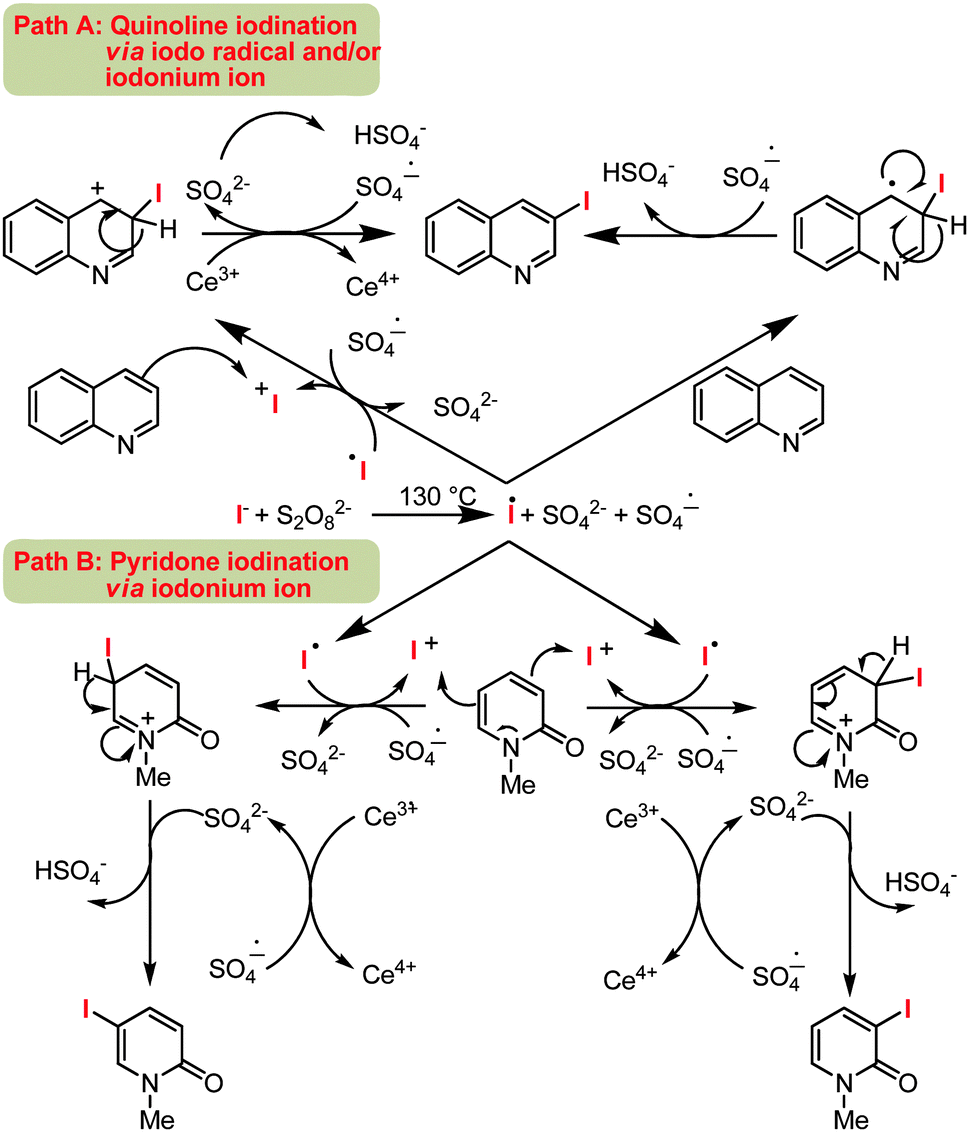 | ||
| Scheme 3 Plausible mechanism. | ||
In conclusion, a variety of heterocycles can be iodinated in a predictable and selective manner using simple reaction conditions. This method is operationally simple and scalable. Due to high demand of heterocyclic iodides, this protocol is expected to find application in industry and academia.
Authors thanks IITB-Monash Research Academy. This activity is supported by SERB-india (EMR/2015/001136). Financial support received from CSIR (A.D.) is gratefully acknowledged.
Notes and references
- (a) P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS PubMed; (b) D. Tilly, F. Chevallier, F. Mongin and P. C. Gros, Chem. Rev., 2014, 114, 1207–1257 CrossRef CAS PubMed; (c) S. Mishra, A. K. Bagdi, M. Ghosh, S. Sinha and A. Hajra, RSC Adv., 2014, 4, 6672–6676 RSC.
- (a) J. F. Bunnett and R. Zahler, Chem. Rev., 1951, 49, 273–412 CrossRef CAS; (b) J. F. Hartwig, Synlett, 2006, 1283–1294 CrossRef CAS; (c) M. R. Crampton, Organic Reaction Mechanisms, Wiley, New York, 2003, p. 275 Search PubMed; (d) A. K. Bagdi, S. Mitra, M. Ghosh and A. Hajra, Org. Biomol. Chem., 2015, 13, 3314–3320 RSC; (e) N. Khatun, S. Guin, S. K. Rout and B. K. Patel, RSC Adv., 2014, 4, 10770–10778 RSC.
- (a) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed; (b) D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450–1460 CrossRef CAS PubMed; (c) I. Popov, S. Lindeman and O. Daugulis, J. Am. Chem. Soc., 2011, 133, 9286–9289 CrossRef CAS PubMed.
- (a) R. Bunevičius, G. Kažanavičius, R. Žalinkevičius and A. J. Prange, N. Engl. J. Med., 1999, 340, 424–429 CrossRef PubMed; (b) S. L. Pimlott and A. Sutherland, Chem. Soc. Rev., 2011, 40, 149–162 RSC; (c) F. Hallouard, N. Anton, P. Choquet, A. Constantinesco and T. Vandamme, Biomaterials, 2010, 31, 6249–6268 CrossRef CAS PubMed; (d) R. H. Seevers and R. E. Counsell, Chem. Rev., 1982, 82, 575–590 CrossRef CAS.
- E. J. Hennessy and S. L. Buchwald, Org. Lett., 2002, 4, 269–272 CrossRef CAS PubMed.
- (a) E. B. Merkushev, Synthesis, 1988, 923–937 CrossRef CAS; (b) T. D. Sheppard, Org. Biomol. Chem., 2009, 7, 1043–1052 RSC; (c) S. K. Santra, A. Banerjee, N. Khatun, A. Samanta and B. K. Patel, RSC Adv., 2015, 5, 11960–11965 RSC; (d) R. K. Chinnagolla, S. Pimparkar and M. Jeganmohan, Chem. Commun., 2013, 49, 3146–3148 RSC; (e) C. B. Tripathi and S. Mukherjee, Angew. Chem., Int. Ed., 2013, 52, 8450–8453 CrossRef CAS PubMed; (f) C. B. Tripathi and S. Mukherjee, Org. Lett., 2014, 16, 3368–3371 CrossRef CAS PubMed.
- (a) T. T. Tsou and J. K. Kochi, J. Org. Chem., 1980, 45, 1930–1937 CrossRef CAS; (b) G. Meyer, Y. Rollin and J. Perichon, Tetrahedron Lett., 1986, 27, 3497–3500 CrossRef CAS; (c) J. H. Clark and C. W. Jones, J. Chem. Soc., Chem. Commun., 1987, 18, 1409–1410 RSC; (d) S. H. Yang, C. S. Li and C. H. Cheng, J. Org. Chem., 1987, 52, 691–694 CrossRef CAS; (e) K.-I. Yamashita, M. Tsuboi, M. S. Asano and K.-I. Sugiura, Synth. Commun., 2012, 42, 170–175 CrossRef CAS PubMed.
- (a) D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Org. Lett., 2006, 8, 2523–2526 CrossRef CAS PubMed; (b) B. Du, X. Jiang and P. Sun, J. Org. Chem., 2013, 78, 2786–2791 CrossRef CAS PubMed; (c) D. Qiu, F. Mo, Z. Zheng, Y. Zhang and J. Wang, Org. Lett., 2010, 12, 5474–5477 CrossRef CAS PubMed; (d) A. Hubbard, T. Okazaki and K. K. Laali, J. Org. Chem., 2008, 73, 316–319 CrossRef CAS PubMed; (e) V. D. Filimonov, N. I. Semenischeva, E. A. Krasnokutskaya, A. N. Tretyakov, H. Y. Hwang and K. W. Chi, Synthesis, 2008, 185–187 CrossRef CAS; (f) E. A. Krasnokutskaya, N. I. Semenischeva, V. D. Filimonov and P. Knochel, Synthesis, 2007, 81–84 CrossRef CAS.
- (a) J. B. Pierce, Z. S. Ariyan and G. S. Ovenden, J. Med. Chem., 1982, 25, 131–136 CrossRef CAS; (b) R. A. Earl and K. P. C. Vollhardt, J. Org. Chem., 1984, 49, 4786–4800 CrossRef CAS; (c) C. A. Mirkin, K. L. Lu, T. E. Snead, G. L. Geoffroy and A. L. Rheingold, J. Am. Chem. Soc., 1990, 112, 2809–2810 CrossRef CAS; (d) T. J. Egan, D. C. Ross and P. A. Adams, FEBS Lett., 1994, 352, 54–57 CrossRef CAS; (e) M. Foley and L. Tilley, Pharmacol. Ther., 1998, 79, 55–87 CrossRef CAS; (f) W. Adam, J. Hartung, H. Okamoto, S. Marquardt, W. M. Nau, U. Pischel, C. R. Saha-Möller and K. Spehar, J. Org. Chem., 2002, 67, 6041–6049 CrossRef CAS PubMed; (g) A. T. Vu, S. T. Cohn, E. S. Manas, H. A. Harris and R. E. Mewshaw, Bioorg. Med. Chem. Lett., 2005, 15, 4520–4525 CrossRef CAS PubMed; (h) F. Surup, O. Wagner, J. V. Frieling, M. Schleicher, S. Oess, P. Muller and S. Grond, J. Org. Chem., 2007, 72, 5085–5090 CrossRef CAS PubMed; (i) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166–187 RSC; (j) V. R. Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488–1508 CrossRef CAS; (k) A. D. Fotiadou and A. L. Zografos, Org. Lett., 2011, 13, 4592–4595 CrossRef CAS PubMed; (l) A. K. Bagdi, S. Santra, M. Rahman, A. Majee and A. Hajra, RSC Adv., 2013, 3, 24034–24037 RSC.
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed; (b) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447–2464 RSC; (c) M. Wasa, B. T. Worrell and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 1275–1277 CrossRef CAS PubMed; (d) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (e) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC; (f) K. Sun, Y. Lv, J. Wang, J. Sun, L. Liu, M. Jia, X. Liu, Z. Li and X. Wang, Org. Lett., 2015, 17, 4408–4411 CrossRef PubMed; (g) K. K. Sharma, D. I. Patel and R. Jain, Chem. Commun., 2015, 51, 15129–15132 RSC.
- (a) E. C. Taylor and A. J. Crovetti, J. Org. Chem., 1954, 19, 1633–1640 CrossRef CAS; (b) R. J. Chambers and A. Marfat, Synth. Commun., 1997, 27, 515–520 CrossRef CAS PubMed; (c) K. C. Nicolaou, K. Namoto, A. Ritzen, T. Ulven, M. Shoji, J. Li, G. D'Amico, D. Liotta, C. T. French, M. Wartmann, K. H. Altmann and P. Giannakakou, J. Am. Chem. Soc., 2001, 123, 9313–9323 CrossRef CAS PubMed; (d) J. G. Ji, T. Li and W. H. Bunnelle, Org. Lett., 2003, 5, 4611–4614 CrossRef CAS PubMed; (e) S. R. Inglis, C. Stojkoski, M. M. Branson, J. F. Cawthray, D. Fritz, E. Wiadrowski, S. M. Pyke and G. W. Booker, J. Med. Chem., 2004, 47, 5405–5417 CrossRef CAS PubMed.
- (a) R. B. Clark, M. S. He, C. Fyfe, D. Lofland, W. J. O’Brien, L. Plamondon, J. A. Sutcliffe and X. Y. Xiao, J. Med. Chem., 2011, 54, 1511–1528 CrossRef CAS PubMed; (b) L. H. Heitman, A. Goblyos, A. M. Zweemer, R. Bakker, T. Mulder-Krieger, J. P. D. van Veldhoven, H. de Vries, J. Brussee and A. P. Ijzerman, J. Med. Chem., 2009, 52, 926–931 CrossRef CAS PubMed; (c) W. Lumeras, F. Caturla, L. Vidal, C. Esteve, C. Balague, A. Orellana, M. Dominguez, R. Roca, J. M. Huerta, N. Godessart and B. Vidal, J. Med. Chem., 2009, 52, 5531–5545 CrossRef CAS PubMed; (d) W. W. Paudler and M. V. Jovanovic, J. Org. Chem., 1983, 48, 1064–1069 CrossRef CAS; (e) A. Petitjean, N. Kyritsakas and J. M. Lehn, Chem. – Eur. J., 2005, 11, 6818–6828 CrossRef CAS PubMed.
- (a) A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 14844–14845 CrossRef CAS PubMed; (b) M. Chen, S. Ichikawa and S. L. Buchwald, Angew. Chem., Int. Ed., 2015, 54, 263–266 CrossRef CAS PubMed.
- L. Li, W.-B. Liu, H.-Y. Zeng, X.-Y. Mu, G. Cosa, Z.-T. Mi and C.-J. Li, J. Am. Chem. Soc., 2015, 137, 8328–8331 CrossRef CAS PubMed.
- S. E. Wengryniuk, A. Weickgenannt, C. Reiher, N. A. Strotman, K. Chen, M. D. Eastgate and P. S. Baran, Org. Lett., 2013, 15, 792–795 CrossRef CAS PubMed.
- H. Hwang, J. Kim, J. Jeong and S. Chang, J. Am. Chem. Soc., 2014, 136, 10770–10776 CrossRef CAS PubMed.
- A. Deb, S. Manna, A. Maji, U. Dutta and D. Maiti, Eur. J. Org. Chem., 2013, 5251–5256 CrossRef CAS PubMed.
- (a) A. Deb, S. Agasti, T. Saboo and D. Maiti, Adv. Synth. Catal., 2014, 356, 705–710 CrossRef CAS PubMed; (b) A. Modak, S. Rana and D. Maiti, J. Org. Chem., 2015, 80, 296–303 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data and spectral data for all compounds. See DOI: 10.1039/c5cc07799k |
| This journal is © The Royal Society of Chemistry 2015 |