
New formulation of old aspirin for better delivery†
Akil A.
Kalathil‡§
,
Anil
Kumar‡§
,
Bhabatosh
Banik‡§
,
Timothy A.
Ruiter§
,
Rakesh K.
Pathak§
and
Shanta
Dhar§
*
NanoTherapeutics Research Laboratory, Department of Chemistry, University of Georgia, Athens, GA 30602, USA. E-mail: shanta@uga.edu
First published on 16th October 2015
Abstract
For better use of cyclooxygenase dependent anti-inflammatory properties and mitochondrial activities of aspirin, new hydrophobic analogues of aspirin were developed and successfully encapsulated in polymeric nanoparticles (NPs). In vivo anti-inflammatory effects of these NPs using a mouse model demonstrated unique properties of an optimized aspirin analogue to inhibit production of pro-inflammatory and enrichment of anti-inflammatory cytokines.
Conditions that include neuro-inflammation, oxidative stress, and mitochondrial injury play different roles in the prognosis of neurodegenerative diseases such as stroke, Alzheimer's disease, Parkinson's disease (PD), Huntington's disease, and amyotrophic lateral sclerosis.1 Although these diseases demonstrate different pathologies, inflammation and oxidative stress are the common players.2 Degradation in mitochondrial health also plays an integral role in overall damage during neuro-degeneration.3 Anti-inflammatory substances such as aspirin and mitochondria-acting antioxidant coenzyme Q10 are described to have potential neuroprotective roles in these diseases.4 Aspirin or acetylsalicylic acid can potentially have a number of roles in neurodegenerative diseases: (i) platelet inhibition through acetylation and prevention of new clots from developing,5 (ii) aspirin can play roles in PD by suppressing formation of dopamine quinone,6 (iii) cyclooxygenase (COX)-independent effect of aspirin on Ca2+ signaling7 for mitochondrial dysfunction related neuro-degeneration.
Current knowledge8 and clinical data9 indicate that aspirin can be an attractive addition to treatment regiments for neurodegenerative diseases. By acknowledging the fact that although few of the nonsteroidal anti-inflammatory drugs such as aspirin can get access to the brain tissue by crossing the tight junctions of the blood brain barrier (BBB), but plasma protein binding activity of this class of molecule limits the effectiveness of such uptake,10 we hypothesized that new hydrophobic analogues of aspirin can be extremely important as aspirin lacks properties required for suitable formulation in a nanoparticle (NP) for better delivery. Further more, gastric toxicity arising from non-specific platelet inhibition by aspirin is a major problem11 and one of the solutions can be slow-release of aspirin at a low dosage.12 Thus, NPs with controlled release properties can provide beneficial manipulation towards pharmacological formulation of aspirin. Additionally, hydrophobic aspirin analogues will help in improving pharmacokinetic (PK) parameters of the generic drug13 as incorporation of new derivatives into a NP system can increase the blood circulation time of the drug when administered by intravenous (i.v.) route in contrast to conventional aspirin administration.
Here we report construction and optimization of new aspirin analogues for their formulation in biodegradable NPs with properties which will allow slow controlled delivery of aspirin in the vicinity of the target tissue and in particular in the mitochondria for possible applications in neuro-degenerative diseases. As a disease model, we investigated utilities of these new aspirin-NP formulations in mice model of inflammation.
Prior to the synthesis of new aspirin derivatives, we assessed whether generic aspirin can be incorporated in the hydrophobic core of biodegradable polymeric NPs. As we would like to target conditions such as mitochondrial dysfunctions associated with oxidative stress, impaired Ca2+ signaling, inflammatory processes demonstrated by brain cells during neurodegenerative processes, we selected a biodegradable poly(lactic-co-glycolic acid)-block-polyethyleneglycol (PLGA-b-PEG) polymer functionalized with a terminal triphenylphosphonium cation (TPP) with significant mitochondrial association properties.14,15 In our continuing effort to evaluate the potential of the targeted NPs (T-NPs) derived from this PLGA-b-PEG–TPP polymer to deliver payload that can work by accessing unique targets at the mitochondria, we first evaluated whether aspirin (Asp) can be incorporated in the T/NT-NPs. Nanoprecipitation of non-targeted PLGA-b-PEG-OH polymer (Fig. S1 and S2, ESI†) or targeted PLGA-b-PEG–TPP polymer (Fig. S3–S5, ESI†) in presence of aspirin afforded low encapsulation efficiency (EE) and percent loading of aspirin inside NT/T-Asp-NPs (Fig. S6, ESI†). Poor encapsulation of aspirin inside the hydrophobic core arises from hydrophilic properties of aspirin. Thus, we hypothesized that construction of hydrophobic analogues which can release aspirin by taking advantages of the hydrolytic agents present in the cellular milieu can be attractive strategy for better delivery of aspirin at the target. This strategy should also result in improved PK and biodistribution (bioD) properties of aspirin when administered in vivo.
Analyses of the properties required for incorporation of aspirin inside hydrophobic core and to increase therapeutic efficacy prompted us to explore the possibility of use of a hydrophobic dendritic platform as the number of required aspirin moieties can easily be tuned.16 The dendritic structure plays important roles in finding optimized structure for aspirin analogue.16b We first developed a first generation [G1] hydrophobic biodegradable dendron with an octyl (Oc) chain connected to two available –OH moieties Oc-[G1]-(OH)2 (4) (Fig. S7–S12, ESI†) for conjugation of two aspirin molecules. This dendron Oc-[G1]-(OH)2 was reacted with aspirin chloride (Fig. S13–S15, ESI†) to generate a hydrophobic dendron Oc-[G1]-(Asp)2 containing two molecules of aspirin linked through cleavable ester bonds (Fig. 1A, Fig. S16–S18, ESI†). Our efforts to encapsulate Oc-[G1]-(Asp)2 in PLGA-b-PEG–TPP polymer to generate T-(Asp)2-NPs and in PLGA-b-PEG-OH polymer to yield NT-(Asp)2-NPs resulted in high loading of the dendron inside the NPs (Table S1, ESI†). However, hydrodynamic diameter of both T/NT-(Asp)2-NPs were ∼200 nm (Table S1 (ESI†), Fig. 1B) which disqualify these NPs to be suitable for mitochondrial association properties.14,15 Next, we increased the number of dendron arms further to increase hydrophobicity of the dendron and constructed Oc-[G2]-(OH)4 (Fig. S19–S24, ESI†) and further conjugation of aspirin resulted in Oc-[G2]-(Asp)4 (Fig. 1A, Fig. S25–27, ESI†) with four aspirin molecules. Incorporation of Oc-[G2]-(Asp)4 into NPs to generate T-(Asp)4-NPs and NT-(Asp)4-NPs indicated sizes below 100 nm and highly positive surface for the T-NPs (Table S2 (ESI†), Fig. 1B). Comparison of NP sizes from these two dendrons indicated that Oc-[G2]-(Asp)4 will be a more appropriate derivative for aspirin delivery. Further, cytotoxicity of T/NT-(Asp)2-NPs and T/NT-(Asp)4-NPs in RAW 264.7 macrophages indicated that the NPs derived from Oc-[G1]-(Asp)2 are relatively more toxic to the cells whereas the NPs from Oc-[G2]-(Asp)4 did not demonstrate any such toxicity up to 100 μM (Fig. 1C). One possible reason for the toxicity of Oc-[G1]-(Asp)2-NPs might be their larger size compared to the Oc-[G2]-(Asp)4 NPs. Transmission electron microscopy (TEM) image analysis of T/NT-(Asp)2-NPs indicated the presence of aggregates with different surface properties (Fig. S28, ESI†). These surface properties may be responsible for the higher toxicity of the T/NT-(Asp)2-NPs.17 Further studies are required to understand the exact mechanism of cytotoxicity of T/NT-(Asp)2-NPs. Based on the size and toxicity of the NPs, we decided to use Oc-[G2]-(Asp)4 for delivery of aspirin using NP platform.
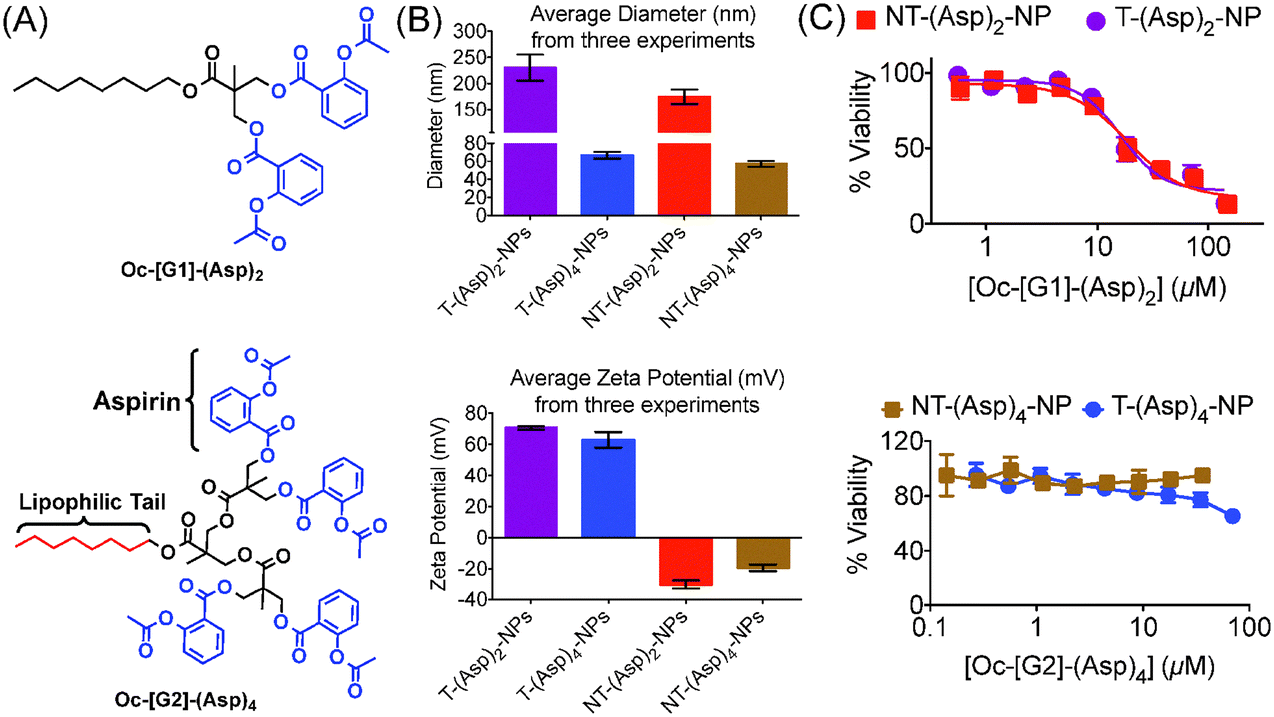 | ||
| Fig. 1 (A) Structures of two newly constructed hydrophobic aspirin derivatives. (B) Analyses of physicochemical properties of the NPs derived from hydrophobic Oc-[G1]-(Asp)2 and Oc-[G2]-(Asp)4 for better delivery. (C) Cytotoxic properties of these NPs in RAW 264.7 macrophages as determined by the MTT assay. | ||
Nanoprecipitation was carried out using 20% feed of Oc-[G2]-(Asp)4 (Fig. 2A) with PLGA-b-PEG–TPP polymer to result in T-(Asp)4-NPs or with PLGA-b-PEG-OH polymer to produce NT-(Asp)4-NPs (Fig. 2B). Control T/NT-empty-NPs were also prepared. Dynamic light scattering (DLS) studies indicated that these NPs have diameter below 100 nm; T-NPs demonstrated high positively charged surface, and the NT-NPs were negatively charged (Fig. 2C). Determination of percent Oc-[G2]-(Asp)4 loading by high performance liquid chromatography (HPLC) indicated loadings of 17 ± 2% for NT and 16.6 ± 0.6% for T NPs, respectively (Fig. 2C). TEM based analyses of the NPs further supported the diameter and confirmed that these spherical NPs are homogeneous (Fig. 2C). Studies suggested that aspirin is an antiplatelet agent that can be effective as an early treatment in acute ischemic stroke and aspirin therapy should be used within 48 h of the initiation of symptoms.18 This made us realize that although controlled release NPs can be invaluable addition to aspirin therapeutic regiments, but the NPs should have release properties where significant portion of aspirin needs to get released in ∼48 h. Investigation of release kinetics of aspirin derivative from T/NT-(Asp)4-NPs under physiological conditions of pH 7.4 at 37 °C demonstrated release of ∼50% Oc-[G2]-(Asp)4 indicating that these NPs might be suitable for aspirin delivery for neuroprotection (Fig. 2D). Aspirin molecules are attached to the dendron structure with aliphatic ester bonds. Thus, in the presence of cellular esterases, the dendron scaffold will release aspirin upon enzymatic hydrolysis.16a Given that the aliphatic esters are more prone to get hydrolyzed as compared to aromatic ester, it should preferentially release aspirin rather than its final metabolite, salicylic acid.
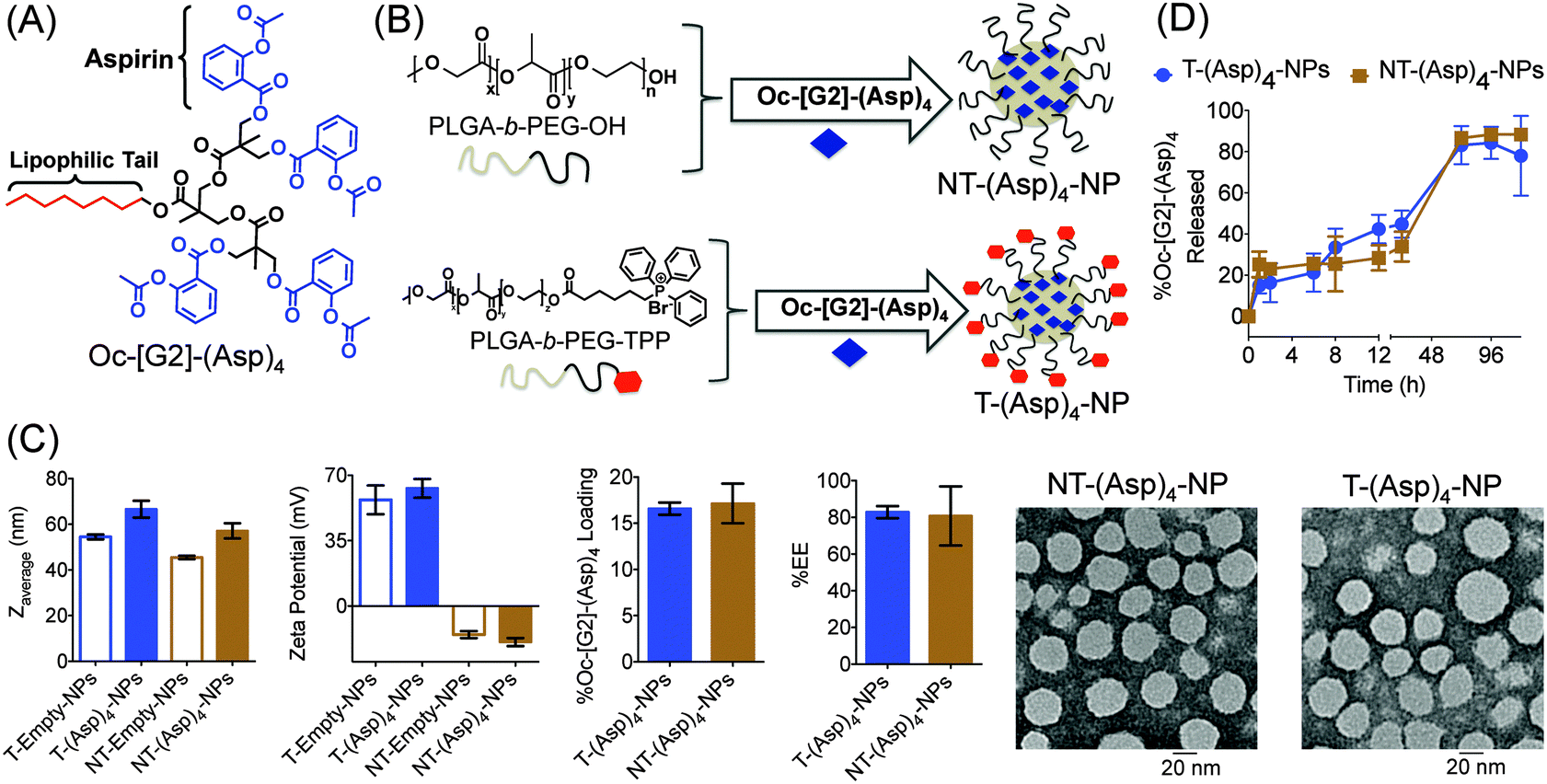 | ||
| Fig. 2 (A) Structure of Oc-[G2]-(Asp)4. (B) Synthesis of T/NT-(Asp)4-NPs from different polymers and Oc-[G2]-(Asp)4. (C) Diameter, zeta potential, percent loading, %EE, and TEM of T/NT-(Asp)4-NPs. (D) Release kinetics of Oc-[G2]-(Asp)4 from T and NT-NPs. | ||
To explore the anti-inflammatory properties of the new aspirin derivative in NP formulation in vivo, we used mice stimulated with lipopolysaccharide (LPS). An earlier study demonstrated that intraperitoneally (i.p.) injected LPS can cause secretion of significant amounts of tumor necrosis factor alpha (TNF-α), which peaks at ∼1.5 h and interleukin-6 (IL-6) at ∼3 h after administration.19 In our studies following similar protocol in C57BL/6 or BALB/c (albino) mice, we observed enhanced levels of TNF-α and IL-6 in the serum after i.p. administration of 100 μg of LPS per animal and the levels peaked at ∼1.5 and ∼3 h for TNF-α and IL-6, respectively (Fig. S29 (ESI†), data shown for C57BL/6 mice). Next, 8 week old BALB/c male mice were administered with saline or aspirin (20 mg kg−1) or NT-(Asp)4-NP (20 mg kg−1 with respect to aspirin) or T-(Asp)4-NP (20 mg kg−1 with respect to aspirin) (Table S3 (ESI†) for characterization of these NPs) by i.v. injections and after 12 h, these animals were subsequently treated with i.p. injected LPS for 1.5 h and 3 h (Fig. 3A). Serum samples were isolated from the treated animals and pro-inflammatory and anti-inflammatory cytokine levels in the serum samples were evaluated by the enzyme-linked immunosorbent assay (ELISA). As seen with C57BL/6 mice, serum TNF-α and IL-6 levels were increased after administration of LPS in BALB/c Albino mice following similar patterns where TNF-α level was peaked at ∼1.5 h and maximum IL-6 level was found at ∼3 h and these levels were significantly higher (P < 0.001) than only saline treated groups (Fig. 3B). Preventative treatment with aspirin (20 mg kg−1) followed by LPS for 1.5 h, TNF-α level was less than LPS alone, but these differences did not reach any statistical significance (LPS vs. aspirin + LPS: non significant for TNF-α, Fig. 3B). Serum samples from the group of animals treated with NT-(Asp)4-NPs prior to LPS treatment for 1.5 h had lower TNF-α than only LPS treated group (P = 0.001–0.01). Most significantly, TNF-α levels from the animals treated with T-(Asp)4-NPs followed by LPS treatment for 1.5 h was drastically reduced compared to only LPS (P < 0.001) (Fig. 3B). Serum TNF-α levels in the T-(Asp)4-NP treated LPS stimulated group was significantly lower than the levels found in the NT-(Asp)4-NP plus LPS treated animals when 1.5 h time point was considered (P < 0.001) (Fig. 3B). Thus, these results indicated that T-(Asp)4-NPs are considerably more effective than free aspirin or NT-(Asp)4-NPs in inhibiting TNF-α production upon LPS stimulation in vivo. Preventative treatment with aspirin, NT-(Asp)4-NPs, or T-(Asp)4-NPs prior to stimulation with LPS for 3 h did not show any significant differences in serum TNF-α levels as this cytokine declined by 3 h (Fig. 3B). In our experimental conditions, the level of IL-6 in only LPS treated samples was significantly increased from that in saline treated group at 1.5 h and the level increased further when LPS treatment was carried out for 3 h. When LPS treatment for 1.5 h was considered, the IL-6 levels were significantly reduced for the groups where preventative treatments were carried out with aspirin, NT-(Asp)4-NPs, or T-(Asp)4-NPs (Fig. 3B). The IL-6 level in the T-(Asp)4-NP treated group was lower than the group administered with NT-(Asp)4-NPs at 1.5 h, however the differences between these two groups did not reach statistical significance (Fig. 3B). These observations indicated that the targeted NP formulation of Oc-[G2]-(Asp)4 is as effective as aspirin in preventing LPS induced IL-6 secretion in vivo. When LPS treatment was carried out for 3 h, only aspirin showed reduced IL-6 levels compared to LPS alone. Empty targeted or non-targeted NPs without aspirin did not show any inflammatory or anti-inflammatory responses under same experimental conditions (Fig. S30, ESI†).
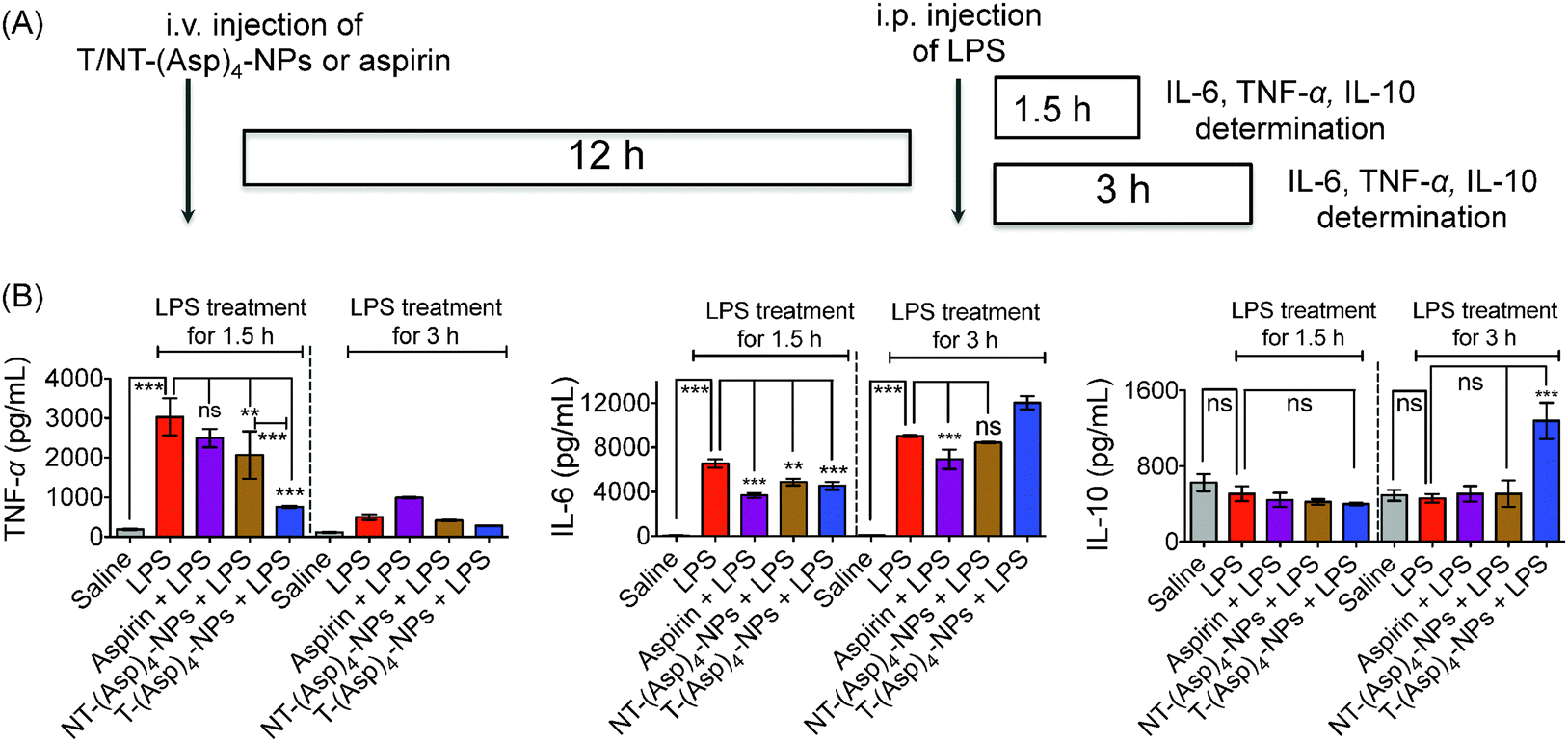 | ||
| Fig. 3 (A) Experimental design for evaluation of anti-inflammatory properties of Oc-[G2]-(Asp)4-NPs under preventative condition using BALB/c albino male mice. (B) Pro-inflammatory TNF-α, IL-6 and anti-inflammatory IL-10 levels in the serum samples of BALB/c albino mice treated with different constructs and LPS. ***: P < 0.001; **: P = 0.001–0.01; ns: non significant. | ||
Anti-inflammatory cytokine, IL-10, determination in the serum samples demonstrated no significant amounts of this cytokine at the 1.5 h of LPS treated samples. However, when 3 h LPS treatment period was considered, a significantly higher level of IL-10 was detected in the serum samples from the animals which were pretreated with T-(Asp)4-NPs prior to LPS stimulation, no other treated group showed such a high IL-10 level (Fig. 3B). The compelling properties of T-(Asp)4-NPs in inhibiting production of pro-inflammatory cytokines and induction of anti-inflammatory IL-10 indicated that the new formulation of aspirin can be an attractive candidate for further exploration for potential activities in inflammation. Detailed mechanistic investigations are needed to understand the effects of aspirin on the mitochondria of cells and possible relation of mitochondrial activity of aspirin with inflammatory properties if there is any.
This work provides first hydrophobic analogue of aspirin which can be loaded inside polymeric NPs efficiently, thus overcoming the disadvantages arising from physicochemical properties of aspirin which do not allow its encapsulation inside the hydrophobic core of NPs. Conjointly, our findings highlighted potential abilities of this new hydrophobic aspirin analogue Oc-[G2]-(Asp)4 encapsulated in a mitochondria targeted NP as a possible therapeutic intervention of the central nervous system inflammation leading to protection against neurodegenerative diseases with inflammatory symptoms.
C56BL/6 (12 weeks old) and BALB/c Albino male mice (8 weeks old) were obtained from Charles River Laboratories and handled in accordance with Animal Welfare Act (AWA), and other applicable federal and state guidelines. All animal work presented here was approved by Institutional Animal Care and Use Committee (IACUC) of University of Georgia.
All statistical analyses were performed using GraphPad Prism software performing a one-way analysis of variance (ANOVA) and nonparametric analyses followed by the Tukey post test.
This work was supported, in whole or in part, by Department of Defense Prostate Cancer Idea award (W81XWH-12-1-0406) and National Institute of Neurological Disorders and Stroke of National Institutes of Health under award number R01NS093314, and by the Office of the Vice President for Research, UGA as a start-up fund to S.D.
References
- S. Amor, F. Puentes, D. Baker and P. van der Valk, Immunology, 2010, 129, 154–169 CrossRef CAS PubMed.
- J. K. Andersen, Nat. Med., 2004, 10(suppl), S18–S25 Search PubMed.
- (a) G. Ashrafi, J. S. Schlehe, M. J. LaVoie and T. L. Schwarz, J. Cell Biol., 2014, 206, 655–670 CAS; (b) H. Chen and D. C. Chan, Hum. Mol. Genet., 2009, 18, R169–176 CrossRef CAS PubMed.
- S. E. Seidl and J. A. Potashkin, Front. Neurol., 2011, 2, 68 Search PubMed.
- A. Undas, K. E. Brummel-Ziedins and K. G. Mann, Blood, 2007, 109, 2285–2292 CrossRef CAS PubMed.
- M. Asanuma, I. Miyazaki, Y. Kikkawa, N. Kimoto, M. Takeshima, S. Murakami and K. Miyoshi, Neurochem. Res., 2012, 37, 1944–1951 CrossRef CAS PubMed.
- (a) A. Szewczyk and L. Wojtczak, Pharmacol. Rev., 2002, 54, 101–127 CrossRef CAS PubMed; (b) Y. Yoshida, I. Singh and C. P. Darby, Acta Neurol. Scand., 1992, 85, 191–196 CrossRef CAS PubMed; (c) T. Tomoda, K. Takeda, T. Kurashige, H. Enzan and M. Miyahara, Liver, 1994, 14, 103–108 CrossRef CAS PubMed.
- B. C. Yan, J. H. Park, B. N. Shin, J. H. Ahn, I. H. Kim, J. C. Lee, K. Y. Yoo, I. K. Hwang, J. H. Choi, J. H. Park, Y. L. Lee, H. W. Suh, J. G. Jun, Y. G. Kwon, Y. M. Kim, S. H. Kwon, S. Her, J. S. Kim, B. H. Hyun, C. K. Kim, J. H. Cho, C. H. Lee and M. H. Won, PLoS One, 2013, 8, e74886 CAS.
- A. S. Zheng, L. Churilov, R. E. Colley, C. Goh, S. M. Davis and B. Yan, JAMA Neurol., 2013, 70, 208–213 CrossRef PubMed.
- (a) J. M. Parepally, H. Mandula and Q. R. Smith, Pharm. Res., 2006, 23, 873–881 CrossRef CAS PubMed; (b) J. P. Courad, D. Besse, C. Delchambre, N. Hanoun, M. Hamon, A. Eschalier, F. Caussade and A. Cloarec, Life Sci., 2001, 69, 1455–1464 CrossRef CAS PubMed.
- C. J. Hawkey and M. J. Langman, Gut, 2003, 52, 600–608 CrossRef CAS PubMed.
- F. L. Lanza, F. K. Chan, E. M. Quigley and G. Practice Parameters Committee of the American College of, Am. J. Gastroenterol., 2009, 104, 728–738 CrossRef PubMed.
- R. O. Day and P. M. Brooks, Br. J. Clin. Pharmacol., 1987, 23, 655–658 CrossRef CAS PubMed.
- S. Marrache and S. Dhar, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16288–16293 CrossRef CAS PubMed.
- (a) S. Marrache, R. K. Pathak and S. Dhar, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10444–10449 CrossRef CAS PubMed; (b) B. Feldhaeusser, S. R. Platt, S. Marrache, N. Kolishetti, R. K. Pathak, D. J. Montgomery, L. R. Reno, E. Howerth and S. Dhar, Nanoscale, 2015, 7, 13822–13830 RSC.
- (a) R. K. Pathak and S. Dhar, J. Am. Chem. Soc., 2015, 137, 8324–8327 CrossRef CAS PubMed; (b) X. Yang, H. Shang, C. Ding and J. Li, Polym. Chem., 2015, 6, 668–680 RSC.
- L. Shang, K. Nienhaus and G. U. Nienhaus, J. Nanobiotechnol., 2014, 12, 5 CrossRef PubMed.
- H. P. Adams, Jr., R. J. Adams, T. Brott, G. J. del Zoppo, A. Furlan, L. B. Goldstein, R. L. Grubb, R. Higashida, C. Kidwell, T. G. Kwiatkowski, J. R. Marler, G. J. Hademenos and A. Stroke Council of the American Stroke, Stroke, 2003, 34, 1056–1083 CrossRef PubMed.
- K. Tateda, T. Matsumoto, S. Miyazaki and K. Yamaguchi, Infect. Immun., 1996, 64, 769–774 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of all experimental methods, synthesis and characterization of different compounds and nanoparticles, and additional data. See DOI: 10.1039/c5cc07316b |
| ‡ A. A. K., A. K., and B. B. contributed equally to this work. |
| § S. D. designed the research and supervised experiments; A. A. K., A. K., B. B.; T. A. R., and R. K. P. performed experiments; A. A. K. and T. A. R. contributed reagents, A. K., B. B., T. A. R., R. K. P., and S. D. analysed the data. The manuscript was written through the contributions of A. A. K., A. K., B. B., R. K. P., and S. D. All authors have given approval to the final version of the manuscript. The authors declare that there are no conflicts of interest. |
| This journal is © The Royal Society of Chemistry 2016 |