
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring water as building bricks in enzyme engineering†
Peter
Hendil-Forssell
a,
Mats
Martinelle
a and
Per-Olof
Syrén
*b
aKTH Royal Institute of Technology, Division of Industrial Biotechnology, AlbaNova University Centre, 106 91 Stockholm, Sweden
bKTH Royal Institute of Technology, Division of Proteomics & Nanobiotechnology, Science for Life Laboratory, 171 21 Stockholm, Sweden. E-mail: per-olof.syren@biotech.kth.se
First published on 25th September 2015
Abstract
A novel enzyme engineering strategy for accelerated catalysis based on redesigning a water network through protein backbone deshielding is presented. Fundamental insight into the energetic consequences associated with the design is discussed in the light of experimental results and computer simulations. Using water as biobricks provides unique opportunities when transition state stabilisation is not easily attained by traditional enzyme engineering.
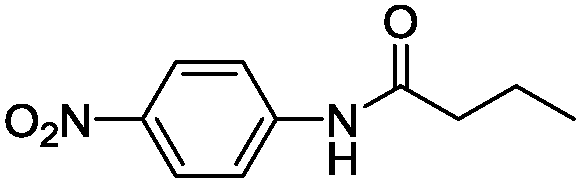
Enzyme engineering constitutes a cornerstone for expanding the catalytic scope of enzymes1 and is pivotal when tailoring biocatalysts to industrial biosynthetic processes.2–4 Enzyme engineering, to create more space or to form favourable enzyme–substrate interactions, has been based on designing small libraries5 that replace key amino acids in and around the active site to attain improved biocatalysts.1,6–13 Furthermore, the repertoire of available building blocks for enzyme design has been expanded through the incorporation of unnatural amino acids into protein scaffolds.14–18 Herein we present an alternative enzyme engineering strategy centred on increasing the water affinity of the protein backbone to achieve efficient transition state (TS) stabilisation through hydrogen bond formation via a water bridge (Fig. 1). In an effort to explore this possibility further we focused on promiscuous amide bond hydrolysis catalysed by esterases for which a missing key hydrogen bond19 in the rate limiting sp2-like nitrogen inversion step19–21 hampers efficient catalysis. This key interaction in amidases and proteases stabilises the TS of nitrogen inversion (Fig. 1a), which is prerequisite to achieve a productive conformation of the lone pair of the reacting nitrogen atom of the amide substrate during catalysis.19,20 We turned our attention to the I189A point mutation in Candida antarctica lipase B (CalB), that to our great surprise was found to afford a 34-fold enhanced promiscuous rate (Δmut-wild
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) typeΔG‡ = −2.1 kcal mol−1, Table 1), despite the fact that the required19 hydrogen bond formation between the scissile NH-group of the amide substrate and the alanine sidechain is not possible. We hypothesized that the substitution affected the solvation of the active site22 to provide enhanced interaction possibilities between the TS and water to allow for accelerated catalysis. Quantum mechanical (QM) calculations in concert with molecular dynamics (MD) simulations revealed that the I189A point mutation reconstituted the missing hydrogen bond through the formation of a short water bridge (Fig. 1c, 2 and Fig. S2, ESI†). Inspired by these facts we decided to evaluate the feasibility and energetic consequences of using water as biological building bricks in enzyme design (Fig. 1c).
typeΔG‡ = −2.1 kcal mol−1, Table 1), despite the fact that the required19 hydrogen bond formation between the scissile NH-group of the amide substrate and the alanine sidechain is not possible. We hypothesized that the substitution affected the solvation of the active site22 to provide enhanced interaction possibilities between the TS and water to allow for accelerated catalysis. Quantum mechanical (QM) calculations in concert with molecular dynamics (MD) simulations revealed that the I189A point mutation reconstituted the missing hydrogen bond through the formation of a short water bridge (Fig. 1c, 2 and Fig. S2, ESI†). Inspired by these facts we decided to evaluate the feasibility and energetic consequences of using water as biological building bricks in enzyme design (Fig. 1c).
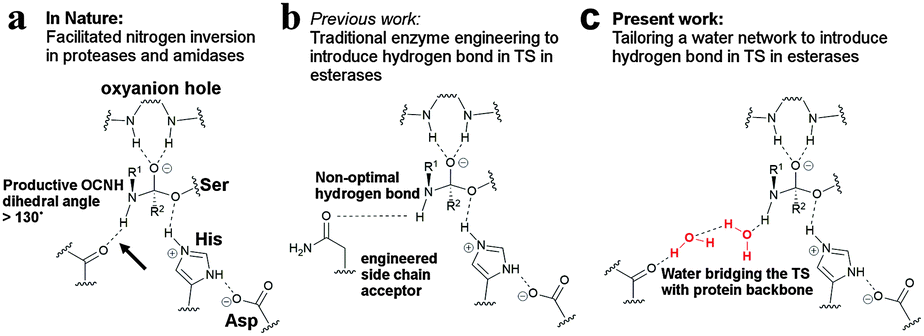 | ||
| Fig. 1 Using water as potential designer tools. (a) Hydrogen bonding to the reacting NH-group of the amide substrate in the TS of nitrogen inversion is essential for efficient catalysis in amidases and proteases. The Ser–His–Asp catalytic machinery of a serine protease is schematically shown together with the oxyanion hole. The key hydrogen bond is shown by the arrow. (b) Traditional enzyme engineering has afforded a direct but weak hydrogen bond in esterases that mimics the key hydrogen bond in amidases.13 (c) A water cluster (red), residing in a precise orientation for interacting with the scissile NH-group in the TS, would reconstitute the missing hydrogen bond during nitrogen inversion in esterases. | ||
| Substrate | Variant | Solvent | ΔH‡ [kcal mol−1] | T × ΔS‡299K [kcal mol−1] | ΔG‡299K [kcal mol−1] | k cat/KM(299K) [M−1 s−1] | Relative reaction specificity ((kcat/KM,amide)/(kcat/KM,ester)) |
|---|---|---|---|---|---|---|---|
|
CalB wt | H2O | 17.3 | −2.45 | 19.8 | 0.022 | 1.0 |
| D2O | 22.8 | 2.20 | 20.6 | 0.0054 | |||
| CalB I189A | H2O | 14.0 | −3.66 | 17.7 | 0.75 | 210 | |
| D2O | 15.0 | −3.08 | 18.1 | 0.36 | |||
|
CalB wt | 12.5 | 1.28 | 11.3 | 37000 |
||
| CalB I189A | H2O | 12.3 | −0.065 | 12.3 | 6100 | ||
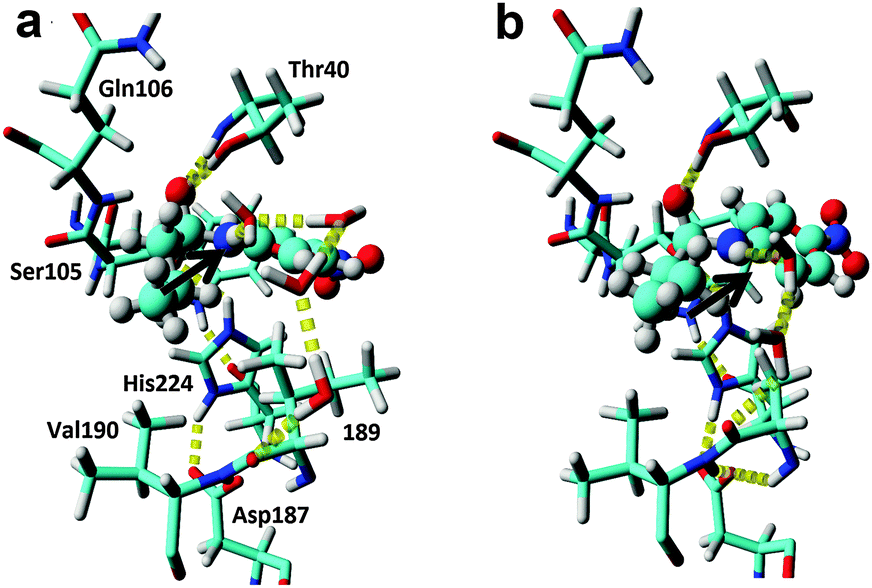 | ||
| Fig. 2 In silico design of a water cluster to achieve efficient TS stabilisation. (a) Snapshot of an MD-simulation of wild type. Typically, larger water clusters involving three to four solvent molecules are necessary to link the substrate NH-group in the TS to the enzyme through hydrogen bond formation. The p-nitrobutyranilide substrate is shown in ball&stick and the key hydrogen bond is shown by the arrow. For the MD-snapshots, the active site entrance (L278, A281 and I285) is not shown for clarity. (b) Snapshot of an MD simulation of the I189A variant. A productive and short water bridge consisting of two water molecules between the TS structure and the deshielded backbone is formed in accordance with the QM calculations (Fig. S2, ESI†). | ||
As amide-based chemistries represent basal organic transformations23 that are of prime interest in the pharmaceutical and biotechnological industrial field,24,25 attempts to enhance promiscuous amidase activities in industrially relevant enzyme scaffolds have previously been performed.13,26,27 Traditional enzyme design has focused on reconstituting the missing hydrogen bond in esterases by engineered acceptor side chains (Fig. 1b).13 Due to spatial constraints in esterases, this has resulted in a tenfold increased promiscuous reaction rate13 by the formation of a direct but non-optimal hydrogen bond in the TS of inversion (Fig. 1b). In order to explore our hypothesis that water could reconstitute a strong hydrogen bond in the TS for the CalB I189A variant, which would explain the enhanced promiscuous activity (Table 1), QM calculations were performed (Fig. S1 and S2, ESI†). Geometry optimization using DFT at the B3LYP/6-31G(d,p) level of theory revealed a perfect hydrogen bond formation between the TS and the exposed backbone in the variant through the participation of two water molecules as building bricks (Fig. S2c, ESI†). In contrast, the key hydrogen bond was lost and the resulting water cluster was found to reside in a catalytically non-productive conformation for the wild-type enzyme (Fig. S2b, ESI†). These results highlights the potential of using enzyme design to increase the water accessibility of polar protein backbone functional groups, that are normally masked by bulky and interacting side chains,28 to allow in theory for the formation of a water bridge between the TS and the protein (Fig. 1c). This strategy would avoid the entropic penalty that would inevitably be associated with restricting the conformational freedom of protein side chains.29 However, enzyme design of a precisely oriented water cluster, which would take full advantage of the capability of water to donate and accept hydrogen bonds, is hampered by our current poor understanding of the influence of solvent dynamics on catalysis.30–34 Furthermore, fixing water molecules inside hydrophobic enzyme cavities could be energetically disfavoured, as compared to water in bulk34,35 and “freezing” the rotational and translational movement of individual and flexible water molecules in transition state structures will be associated with high entropic costs. Thus, in order to shed additional light on the water cluster geometries beyond the “frozen” picture provided by the QM calculations, MD simulations were performed on the second tetrahedral intermediate during acylation (Fig. 2a and b), which was used to represent the TS for nitrogen inversion.20 The O–C–N–H dihedral angle was not held fixed during the MD simulations in order to sample both productive nitrogen inversions (Fig. 1a, dihedrals >130°) and unproductive transition state structures (a threshold value of 130° for the O–C–N–H dihedral was chosen as proteases have been found to display corresponding angles that exceed 130° during their facilitated inversion process).19 Monitoring of all possible bridging interactions between the scissile NH-group and explicit water and protein was performed during 100 ns MD for the wild-type enzyme (Fig. S3a, ESI†). From the MD simulations, that were run in duplicates, it can be concluded that the active site of the esterase enzyme is solvated (Table S1, ESI†). Complex water clusters in the wild type (Fig. S3a, ESI†) provided a hydrogen bond to the scissile NH-group of the substrate in 9% of the analysed 40000 snapshots (Table S2, ESI†). However, only a minority of the total snapshots (0.05%) concomitantly displayed a hydrogen bonded water molecule and an O–C–N–H dihedral angle larger than 130°, which would correspond to a productive TS for nitrogen inversion (Fig. S4, ESI†). Furthermore, for the wild-type enzyme the water H-bond acceptor was typically involved in water bridges involving three or four water molecules to enzyme acceptors at positions T40, D134, Q157, I189 and A281 (Fig. S3a, ESI†). “Freezing” such large clusters of solvent molecules in the enzyme active site during TS formation is expected to be associated with severe entropic penalties. Furthermore, these water molecules were on average stabilised with only 2 hydrogen bonds, (Tables S1 and S2, ESI†) in contrast to 3.6 in bulk.30 Taken together, these facts indicate a possible stabilisation of the TS for nitrogen inversion for wild type through the formation of a key hydrogen bond to a water molecule (Fig. S2, ESI†), which could explain the low promiscuous amidase activity displayed by the wild-type enzyme.
Next, duplicate MD-simulations of the I189A variant were performed to evaluate additional stabilisation of the key water acting as hydrogen bond acceptor in the TS. It was found that the probability of H-bond formation between the scissile NH-group and a water molecule was very similar to that of the wild-type enzyme. Interestingly, from the MD-simulations it was found that the active site contained one additional water molecule as compared to wild type (Table S1, ESI†), presumably as a consequence of the additional cavity formed. Moreover, profound differences in the water clusters that bridged the reacting NH-group in the modelled TS with the protein were detected (Fig. S3b, ESI†). Importantly, the large clusters consisting of 3–4 water molecules, and that were interacting with D134, Q157 and A281 in the wild type, were replaced in favour of a short water bridge to the backbone at position 189 in the mutated enzyme (Fig. S3b, ESI†), which would be in accordance with the QM results (Fig. S2c, ESI†). The probability of forming such a short water bridge to the backbone of 189 increased from 0.5% in the wild-type enzyme to 3.6% in the Ala variant (Table S3, ESI†). Moreover, the altered water patterns were associated with a significant shift in the O–C–N–H dihedral angle population towards higher values for the I189A variant, as compared to wild type (Fig. S5, ESI†). The I189A variant displayed a relative probability increase of one order of magnitude to display dihedral angles that would correspond to productive transition state structures. Furthermore, the introduced mutation increased the quality of the hydrogen bond network as each active site water displayed slightly more hydrogen bonds on average compared to wild type (Tables S1 and S2, ESI†).
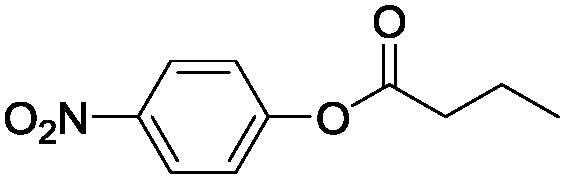
Taken together, the QM and MD results support a more exposed backbone for the I189A variant that is more prominent in stabilising a water-mediated key hydrogen bond via short water clusters, which would be less entropically disfavoured as opposed to freezing larger networks of water. Furthermore, the QM calculations revealed that I189 constituted a “hotspot”, as it displayed a backbone carbonyl that resided in proximity of the reacting amide bond and on the opposite side compared to the catalytic Ser (Fig. S2a, ESI†), corresponding to the antiperiplanar spatial arrangement of the hydrogen bond acceptor found in amidases (Fig. 1a).20 Hence additional functionalization/defunctionalisation at this key position would give insight into the role of water as potential building bricks in enzyme design. For this purpose four additional variants were designed and analysed both computationally and experimentally (Fig. S3c, S6, S7 and Tables S4, S5, ESI†). A shorter MD simulation (50 ns) on the I189V variant revealed a lower relative probability (0.1%) of generating a short water bridge between the TS and the protein compared to that of the wild-type enzyme (0.5%, Table S3 and Fig. S3c, ESI†). This would be in accordance with a protected backbone and with the experimental observations that the I189V variant displays slightly lower amidase activity compared to that of wild type (Table S4, ESI†).13 Computational analysis of the I189G variant (representing maximal deshielding) unravelled the formation of a water bridge consisting of a single water molecule (Fig. S7a, ESI†) with an associated relative probability of 0.5%, which is close to the value displayed by wild type for the formation of a cluster consisting of two waters. Kinetic experiments confirmed that the I189G variant displayed a 6-fold higher absolute amidase activity compared to that of wild type in accordance with a favourable lower entropic cost of freezing one water molecule in the I189G variant. Remarkably, the I189G variant displayed a shift in the amidase over esterase reaction specificity of 84-fold at 37 °C (Tables S4 and S5, ESI†). Interestingly, the additional functionalization displayed by the I189S and I189N variants was associated with complex and highly-ordered transition state structures for which larger water clusters bridged the introduced side chain and TS, concomitantly with the formation of short clusters to the deshielded backbone (Fig. S6 and S7, ESI†). All analysed variants displayed an experimentally determined promiscuous reaction rate that was correlated to the probability to form a short water bridge between the TS of inversion and the protein backbone (see supplementary note on water patterns in ESI,† and Fig. S7 for representative water patterns in TS). Moreover, the low promiscuous rate displayed by I189S (Table S4, ESI†) stresses that cavity formation did not facilitate nitrogen inversion per se. Visual inspection demonstrated that rotation around the C–N bond of the substrate, to circumvent nitrogen inversion, was not feasible (Fig. S8, ESI†). To shed fundamental insight into the energetic consequences of a reconfigured water cluster, the absolute specificity (kcat/KM) of wild type and the I189A variant towards the hydrolysis of p-nitrophenyl butyrate (ester) and p-nitrobutyranilide (amide) was determined at different temperatures in both water and heavy water (Table 1). The 34-fold absolute rate enhancement for amide bond hydrolysis displayed by I189A was associated with a six-fold drop in esterase activity as compared to that of wild type at 26 °C (Table 1). As an ester oxygen has two lone pairs, there is no need for inversion during ester hydrolysis and hence no rate enhancement is expected upon introducing hydrogen bonding acceptors in close vicinity of the substrate.13,21 The overall effect of the point mutation was a 210-fold increase in the relative amidase over esterase reaction specificity as compared to that of wild type at 26 °C (Table 1). The striking change in reaction specificity is not associated with structural rearrangements as confirmed by circular dichroism (Fig. S11 and Table S6, ESI†). In H2O, the 34-fold accelerated amide bond hydrolysis rate displayed by the I189A variant (corresponding to Δmut-wildtypeΔG‡ = −2.1 kcal mol−1) was associated with a large decrease of 3.3 kcal mol−1 in activation enthalpy (Δmut-wildtypeΔH‡), in accordance with stabilization of the TS for nitrogen inversion by enhanced hydrogen bond formation. Simultaneously there was a decrease in the activation entropy (T × Δmut-wildtypeΔS‡) of 1.2 kcal mol−1 disfavouring catalysis, which would be in accordance with “freezing” water molecules in the transition state.30,35 For ester hydrolysis, that proceed without inversion,20 the six-fold decrease in absolute activity displayed by the I189A variant was associated with a disadvantageous change in the entropic state function (i.e. T × Δmut-wildtypeΔS‡ = −1.3 kcal mol−1), in analogy to the value found for amide bond hydrolysis (i.e. −1.2 kcal mol−1). In contrast, the activation enthalpy displayed by the variant was essentially unchanged for ester hydrolysis (Table 1, bottom) in accordance with catalysis without inversion and hydrogen bond stabilisation. These results are aligned with the fixation of a small water cluster in the TS for both ester and amide bond hydrolysis catalysed by the I189A variant. Furthermore, the significantly lower activation enthalpy displayed by the I189A variant in D2O compared to that of wild type is in accordance with increased hydrogen bonding strength in heavy water (Table 1, see supporting note (ESI†) on solvent isotope effects).36 The 5.3 kcal mol−1 lower activation entropy for I189A compared to that of the wild type in D2O at 26 °C, is in accordance with the well-known fact of the higher entropic costs of freezing a free D2O molecule compared to H2O.36
Water is fundamental for the chemistry of life.30 It is known that explicit water molecules play central roles in chemistry37 and in biology by providing a driving force in ligand association,34,35,38 and for specificity39 and activity.22,31,32,39 Herein, thermodynamical experimental data in concert with computer modelling suggests that water constitute potential building blocks in enzyme design, as a deshielded backbone accelerates catalysis through the formation of a short water bridge between the productive TS and the protein backbone. The experimentally observed resulting change in absolute specificity of 34-fold displayed by CalB I189A, is to the best of our knowledge the highest promiscuous amidase activity obtained this far. The tailored water network that facilitates stabilisation of the TS for nitrogen inversion was associated with an entropic penalty of 1.2 kcal mol−1, which is much less than the cost for freezing one water molecule in a salt crystal.40 Furthermore, a linear enthalpy–entropy compensation plot (Fig. S10, ESI†) indicates that the reconfigured water network did not alter the reaction mechanism. Hence, using water as biobricks could constitute a promising strategy when transition state stabilisation is not easily attained by traditional enzyme engineering.
The Swedish Research Council (VR) is greatly acknowledged for financial support of this work by a young investigator grant #621-2013-5138. The PDC Center for High Performance Computing at the KTH Royal Institute of Technology and the Protein Science Facility at SciLifeLab are greatly acknowledged. Author contributions: P.H.F. planned and performed laboratory experiments and helped to write the manuscript. M.M. acted as consultant. P.O.S. planned experiments, performed simulations and analysis and wrote the manuscript.
Notes and references
- M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- B. M. Nestl, S. C. Hammer, B. A. Nebel and B. Hauer, Angew. Chem., Int. Ed., 2014, 53, 3070–3095 CrossRef CAS PubMed.
- N. J. Turner, Nat. Chem. Biol., 2009, 5, 567–573 CrossRef CAS PubMed.
- S. Lutz, Curr. Opin. Biotechnol., 2010, 21, 734–743 CrossRef CAS PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- G. P. Horsman, A. M. F. Liu, E. Henke, U. T. Bornscheuer and R. J. Kazlauskas, Chem. – Eur. J., 2003, 9, 1933–1939 CrossRef CAS PubMed.
- H. Jochens and U. T. Bornscheuer, ChemBioChem, 2010, 11, 1861–1866 CrossRef CAS PubMed.
- Q. Wu, P. Soni and M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 1872–1881 CrossRef CAS PubMed.
- S. C. Hammer, A. Marjanovic, J. M. Dominicus, B. M. Nestl and B. Hauer, Nat. Chem. Biol., 2015, 11, 121–126 CrossRef CAS PubMed.
- P. S. Coelho, Z. J. Wang, M. E. Ener, S. A. Baril, A. Kannan, F. H. Arnold and E. M. Brustad, Nat. Chem. Biol., 2013, 9, 485–487 CrossRef CAS PubMed.
- H. Renata, Z. J. Wang and F. H. Arnold, Angew. Chem., Int. Ed., 2015, 54, 3351–3367 CrossRef CAS PubMed.
- P.-O. Syren, P. Hendil-Forssell, L. Aumailley, W. Besenmatter, F. Gounine, A. Svendsen, M. Martinelle and K. Hult, ChemBioChem, 2012, 13, 645–648 CrossRef CAS PubMed.
- L. Wang, J. Xie and P. G. Schultz, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 225–249 CrossRef CAS PubMed.
- Z. E. Reinert, G. A. Lengyel and W. S. Horne, J. Am. Chem. Soc., 2013, 135, 12528–12531 CrossRef CAS PubMed.
- N. Morikubo, Y. Fukuda, K. Ohtake, N. Shinya, D. Kiga, K. Sakamoto, M. Asanuma, H. Hirota, S. Yokoyama and T. Hoshino, J. Am. Chem. Soc., 2006, 128, 13184–13194 CrossRef CAS PubMed.
- M. Amaro, J. Brezovsky, S. Kovacova, J. Sykora, D. Bednar, V. Nemec, V. Liskova, N. P. Kurumbang, K. Beerens, R. Chaloupkova, K. Paruch, M. Hof and J. Damborsky, J. Am. Chem. Soc., 2015, 137, 4988–4992 CrossRef CAS PubMed.
- I. N. Ugwumba, K. Ozawa, Z.-Q. Xu, F. Ely, J.-L. Foo, A. J. Herlt, C. Coppin, S. Brown, M. C. Taylor, D. L. Ollis, L. N. Mander, G. Schenk, N. E. Dixon, G. Otting, J. G. Oakeshott and C. J. Jackson, J. Am. Chem. Soc., 2011, 133, 326–333 CrossRef CAS PubMed.
- P.-O. Syren and K. Hult, ChemCatChem, 2011, 3, 853–860 CrossRef CAS PubMed.
- P.-O. Syren, FEBS J., 2013, 280, 3069–3083 CrossRef CAS PubMed.
- P.-O. Syren, F. Le Joubioux, Y. Ben Henda, T. Maugard, K. Hult and M. Graber, ChemCatChem, 2013, 5, 1842–1853 CrossRef CAS PubMed.
- M. Pavlova, M. Klvana, Z. Prokop, R. Chaloupkova, P. Banas, M. Otyepka, R. C. Wade, M. Tsuda, Y. Nagata and J. Damborsky, Nat. Chem. Biol., 2009, 5, 727–733 CrossRef CAS PubMed.
- J. Aube, Angew. Chem., Int. Ed., 2012, 51, 3063–3065 CrossRef CAS PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CAS.
- S. Hackenschmidt, E. J. Moldenhauer, G. A. Behrens, M. Gand, I. V. Pavlidis and U. T. Bornscheuer, ChemCatChem, 2014, 6, 1015–1020 CrossRef CAS PubMed.
- Y. Nakagawa, A. Hasegawa, J. Hiratake and K. Sakata, Protein Eng., Des. Sel., 2007, 20, 339–346 CrossRef CAS PubMed.
- A. S. Holehouse, K. Garai, N. Lyle, A. Vitalis and R. V. Pappu, J. Am. Chem. Soc., 2015, 137, 2984–2995 CrossRef CAS PubMed.
- S.-R. Tzeng and C. G. Kalodimos, Nature, 2012, 488, 236–240 CrossRef CAS PubMed.
- P. Ball, Chem. Rev., 2008, 108, 74–108 CrossRef CAS PubMed.
- P. W. Snyder, J. Mecinoviv, D. T. Moustakas, S. W. Thomas, III, M. Harder, E. T. Mack, M. R. Lockett, A. Heroux, W. Sherman and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17889–17894 CrossRef CAS PubMed.
- P.-O. Syren, S. C. Hammer, B. Claasen and B. Hauer, Angew. Chem., Int. Ed., 2014, 53, 4845–4849 CrossRef CAS PubMed.
- L. Wang, E. A. Althoff, J. Bolduc, L. Jiang, J. Moody, J. K. Lassila, L. Giger, D. Hilvert, B. Stoddard and D. Baker, J. Mol. Biol., 2012, 415, 615–625 CrossRef CAS PubMed.
- R. Abel, T. Young, R. Farid, B. J. Berne and R. A. Friesner, J. Am. Chem. Soc., 2008, 130, 2817–2831 CrossRef CAS PubMed.
- T. Young, R. Abel, B. Kim, B. J. Berne and R. A. Friesner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 808–813 CrossRef CAS PubMed.
- C. G. Swain and R. F. W. Bader, Tetrahedron, 1960, 10, 182–199 CrossRef CAS.
- J. A. Byers and T. F. Jamison, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16724–16729 CrossRef CAS PubMed.
- J. Michel, J. Tirado-Rives and W. L. Jorgensen, J. Am. Chem. Soc., 2009, 131, 15403–15411 CrossRef CAS PubMed.
- S. Matsuoka, S. Sugiyama, D. Matsuoka, M. Hirose, S. Lethu, H. Ano, T. Hara, O. Ichihara, S. R. Kimura, S. Murakami, H. Ishida, E. Mizohata, T. Inoue and M. Murata, Angew. Chem., Int. Ed., 2015, 54, 1508–1511 CrossRef CAS PubMed.
- J. D. Dunitz, Science, 1994, 264, 670 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc07162c |
| This journal is © The Royal Society of Chemistry 2015 |