
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
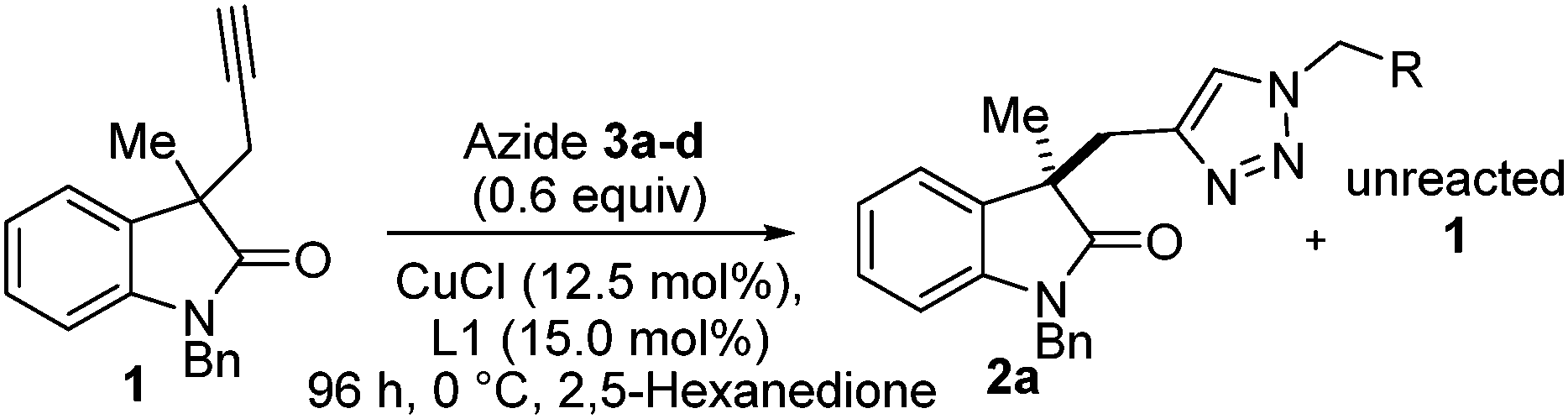
Kinetic resolution of alkyne-substituted quaternary oxindoles via copper catalysed azide–alkyne cycloadditions†
William D. G.
Brittain
a,
Benjamin R.
Buckley‡
*b and
John S.
Fossey‡
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, West Midlands B15 2TT, UK. E-mail: j.s.fossey@bham.ac.uk
bDepartment of Chemistry, Loughborough University, Loughborough, Leicestershire LE11 3TU, UK. E-mail: b.r.buckley@lboro.ac.uk
First published on 27th August 2015
Abstract
The synthesis and kinetic resolution of quaternary oxindoles through copper catalysed azide–alkyne cycloadditions is presented. Selectivity factors (s) up to 22.1 ± 0.5 are reported. Enantioenriched alkynes and triazoles were obtained in ≥80% enantiomeric excess (e.e.).
Click chemistry was first outlined in 2001 by K. B. Sharpless and co-workers, the copper catalysed azide alkyne cycloaddition (CuAAC) has lived up to Sharpless' accolade as “the cream of the crop” in this area.1 The CuAAC reaction predictably delivers 1,4 substituted triazoles in good yields, from the reaction of terminal alkynes with organic azides in the presence of a Cu(I) catalyst. Despite the ubiquity of the CuAAC reaction there are relatively few reports of asymmetric variants.2 Yet enantioenriched triazoles and alkynes are potentially important in many areas of chemistry and biology.3
Kinetic resolution (KR) of a racemic mixture takes advantage of a difference in the rate of the reaction of either enantiomer through diastereomeric interactions.4 Fu and co-workers have championed non-enzymatic catalytic methods for the KR of secondary alcohols and other substrates.5 In this area recent advances have built on Fu's findings and resolution of chiral alcohols is a relatively mature field.6 A particular advantage of KR is that it is possible to obtain high e.e.s of both unreacted starting materials and products dependent upon the selectivity factor (s) and the conversion. Kagan showed that for a given KR it is relatively straight forwards to relate conversion and starting material e.e. to selectivity factor. KR of chiral alkynes or azides through triazole formation is one strategy open to exploitation to access enantioenriched triazoles and starting materials, thus enabling recovery of non-racemic alkynes which could be further derivatised.
To the best of our knowledge there are no reports of successful KRs of alkynes via the CuAAC reaction. Kinetic resolution of terminal alkynes is therefore of importance to the overall development of stereoselective click chemistry. Research into asymmetric click methodology thus far has been mainly focused on desymmetrisation and only a single successful report on KR (of azides) has been published.
Resolution of racemic azides via the CuAAC reaction has been achieved by Meng et al.7 using a Cu–PyBox catalyst,8 selectivity factors (s) up to s = 8 where obtained.9 Meng et al. were unable to kinetically resolve racemic terminal alkynes (s = 1).7 A range of five different terminal alkynes were tested and no enantiomeric discrimination was observed whatsoever with any compounds tried. They accounted for this observation using a mononuclear transition state model. However, more recently evidence has emerged that Cu–PyBox systems might not always follow a mononuclear arrangement. Panera et al. reported X-ray crystal structures of binuclear phenyl and isopropyl PyBOX copper(I) chloride complexes, and used these solid state structures to rationalise the stereochemical outcome of their asymmetric catalytic synthesis of propargylamines.14 It struck us that this binuclear PyBox model could have synergy with the binuclear model for the CuAAC, proposed by Fokin et al.15 and Kuang et al.16 Work by Jin et al. and Makarem et al. has provided strong evidence to support this catalytic model with stable dinuclear copper acetylide intermediates being successfully crystallised.17
Our team has a long-standing interest in quaternary oxindoles,18 and a relevant report on desymmetrisation,2a prompted us to begin investigation of their potential as substrates for catalytic kinetic resolution. Oxindole derivatives (Fig. 1) can possess potent biological activity as calcium channel blockers,19 anti-angiogenics,20 antitumor agents21 and analgesics.19,22 For example, the antitumor agent I, the natural products donaxaridine II, dioxibrassinine III and the popular natural product target gelsamine IV (Fig. 1). Therefore, efficient access to nonracemic quaternary oxindoles is of considerable interest to the medicinal chemistry23 and veterinary science24 communities. Not only is access to non-racemic triazoles of interest but access to high e.e. alkynes is of importance. Access to enantioenriched all carbon quaternary stereogenic centres bearing alkyne functionality is demanding,25 and alkynes are a versatile synthetic handle capable of undergoing a diverse array of transformations.26 Access to enantioenriched alkynes therefore could lead to a plethora of stereocontrolled derivatives. In order to test whether kinetic resolution of alkyne appended quaternary oxindoles is possible (generically represented in Scheme 1a) we prepared compound 1. Propargylation of 3-methyl-2-oxindole (see ESI†), followed by N-benzylation gave racemic 1 (Scheme 1b), our substrate for KR.
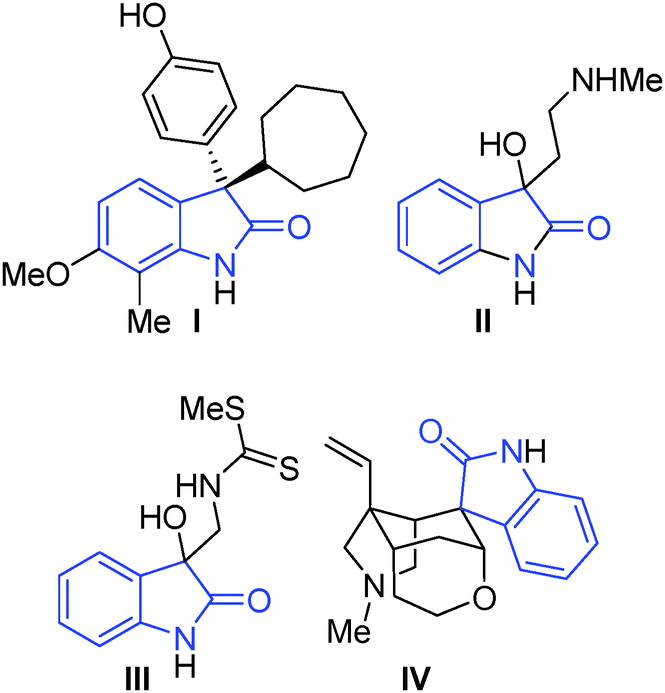 | ||
| Fig. 1 Exemplar biologically active quaternary C-3 oxindoles; I: antitumor agent;10II: donaxaridine;11III: dioxibrassinine;12IV: gelsemine.13 | ||
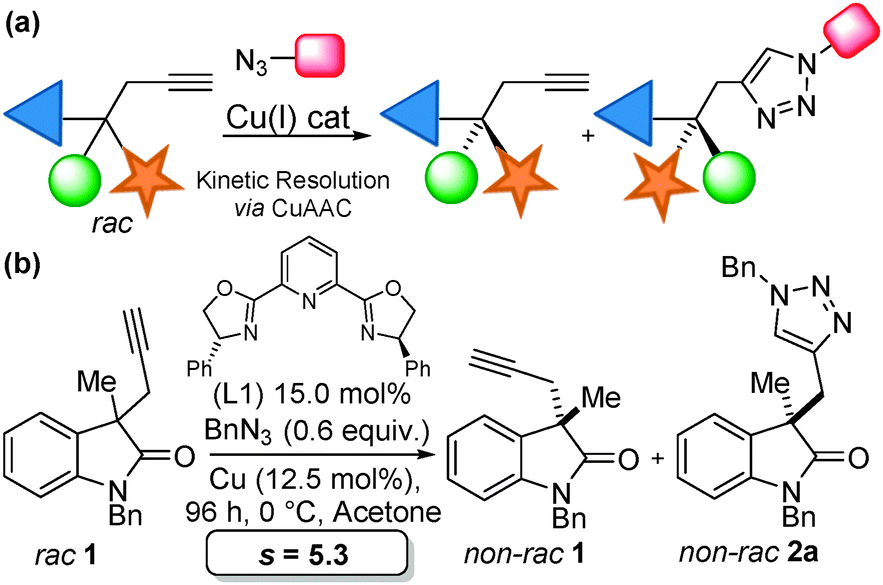 | ||
| Scheme 1 (a) General scheme for the CuAAC KR strategy; (b) application of KR to alkynyl C-3 quaternary oxindoles. | ||
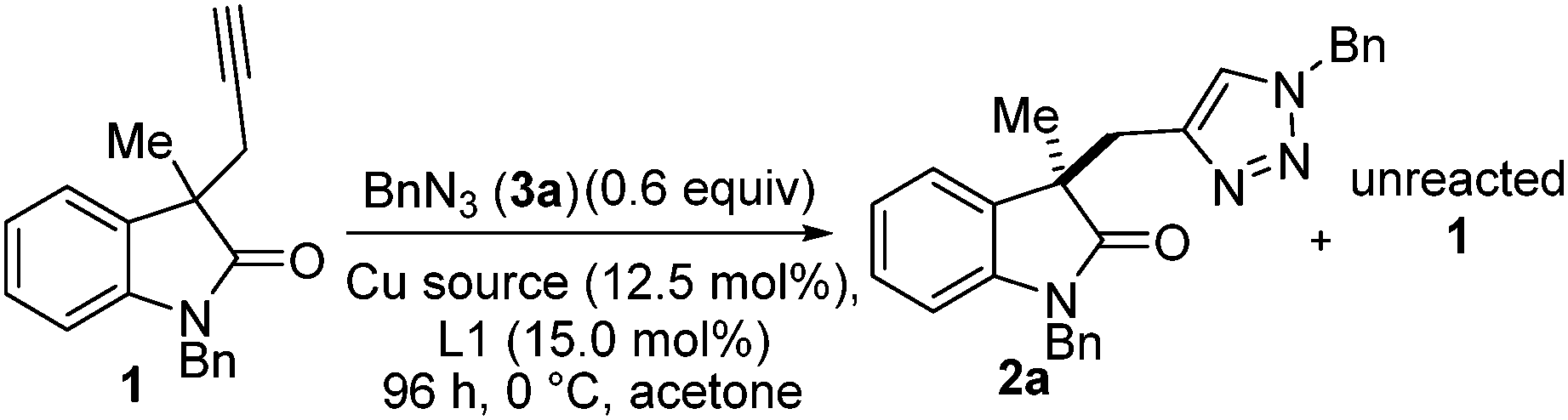
To our delight, an initial probe reaction of racemic 1 with benzyl azide, in acetone under control of a combination of Cu(I)Cl and PhPyBOX (L1) (12.5 and 15 mol% respectively) gave a selectivity of s = 5.3. Conversion was determined by examination of proton NMR spectrums and e.e. by HPLC analysis (see ESI† for details). Optimisation of the reaction conditions was then explored. A series of readily available copper sources were probed, including Cu(I) Cu(II) and Cu(0) species, under standard conditions of 0.6 equivalents of benzyl azide (3b), 12.5 mol% of copper source and 15 mol% of PhPyBox ligand (L1) at 0 °C for 96 h (Table 1). Of copper sources tested, our first choice, Cu(I)Cl was confirmed as the superior choice selectivity factor (Table 1, entry 1). Cu(I)Br gave a lower conversion and selectivity factor (Table 1, entry 2); Cu(I)I offered improved conversion but selectivity factor was again compromised (Table 1, entry 3). Of the other copper sources tried Cu(I)OTf, Cu(II)(OAc)2 and CuSO4 combined with NaAsc (Table 1, entries 4, 6 and 7 respectively) gave approximately stoichiometric conversion with respect to catalyst loading. Cu(I)OAc, Cu(II)OTf2 and Cu(0) NaAsc (Table 1, entries 5, 8 and 9 respectively) did not deliver any triazole-containing products. Increasing the reaction temperature did not improve the reaction outcomes and Cu(I)Cl was selected as the copper source of choice for further optimisation studies.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Copper Source | Conv.a (%) | e.e. SMb (%) | e.e. 2ab (%) | Selectivity factor (s)c |
| a Conversion determined by inspection of 1H NMR spectra (see ESI). b e.e. of recovered starting material (HPLC). c s = ln[(1 − c)(1 − ee)]/ln[(1 − c)(1 + ee)]. | |||||
| 1 | CuCl | 37 | 34 | 43 | 5.3 |
| 2 | CuBr | 14 | 8 | 8 | 3.2 |
| 3 | CuI | 51 | 26 | 29 | 2.1 |
| 4 | Cu(OTf)·toluene 0.5 | 10 | 3 | 16 | 1.8 |
| 5 | Cu(OAc) | 0 | 0 | — | — |
| 6 | Cu(OAc)2 | 8 | 4 | 57 | 2.8 |
| 7 | CuSO4, NaAsc | 13 | 1 | — | — |
| 8 | Cu(OTf)2 | 0 | 0 | — | — |
| 9 | Cu metal | 0 | 0 | — | — |
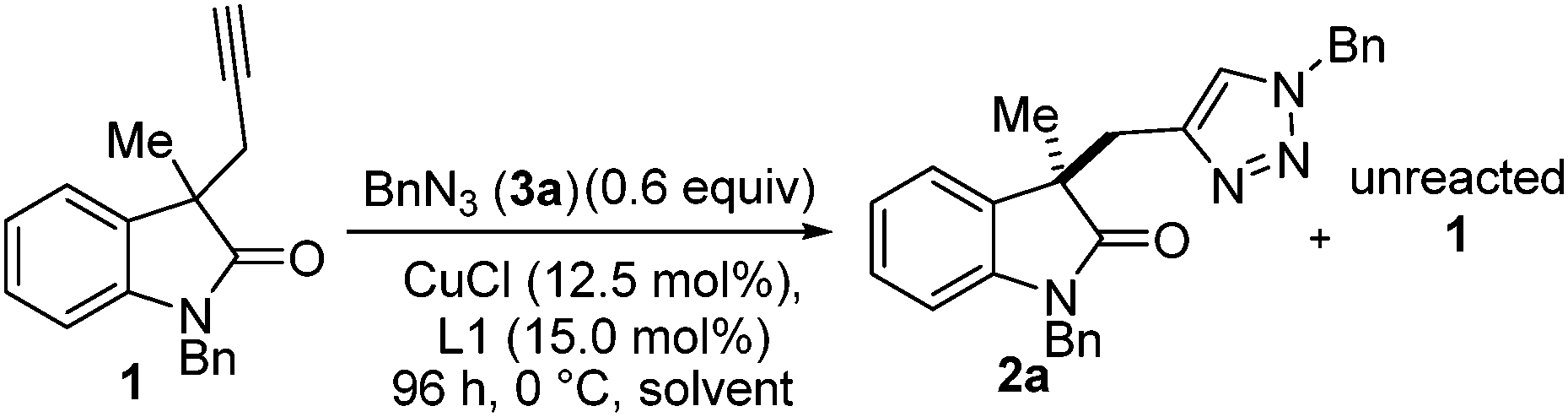
Next, the choice of solvent was investigated (Table 2), acetone had already been shown to give a promising selectivity and is restated in Table 2 (entry 1) to aid comparison. Surprisingly 2-butanone was ineffective (Table 2, entry) giving no conversion. Since Zhou et al. had already reported dicarbonyl containing solvents were effective for a related desymmetrisation reaction we also included this class of solvent in our screening.2a,27 2,5-Hexanedione (Table 2, entry 3) gave a huge jump in selectivity factor (s = 22.1 ± 0.5) and good conversion (42%), 2,3-butanedione (Table 2, entry 4) gave poorer conversion and poorer selectivity (34% and s = 2.5 respectively). A mixture of acetone and 2,5-hexanedione (Table 2, entry 5) gave 61% conversion (against 0.6 equiv. of azide, i.e. full conversion) but selectivity factor was also poor (s = 3.5). THF gave a promising selectivity factor of s = 8.7 (Table 2, entry 6), and all other solvents tried gave inferior results (Table 2, entries 7–12). We speculated that 2,5-hexanedione may be acting as a ligand for copper but use in substoichiometric amounts (equivalent to catalyst loading) had a negative effect on the selectivity. During the course of this optimisation study the order of addition was found to be crucial to obtaining reproducible results. Importantly, the alkyne must be added to a solution of in situ formed catalyst at room temperature before cooling to 0 °C (see ESI† for full details).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Conv.a (%) | e.e. SMb (%) | e.e. 2ab (%) | Selectivity factor (s)c |
| a Conversion determined by inspection of 1H NMR spectra (see ESI) b e.e. of recovered starting material (HPLC) c s = ln[(1 − c)(1 − ee)]/ln[(1 − c)(1 + ee)] d Average of three s = 22.1 ± 0.5, best unique case s = 23.2. | |||||
| 1 | Acetone | 37 | 34 | 43 | 5.3 |
| 2 | 2-Butanone | 0 | — | — | — |
| 3 | 2,5-Hexanedione | 42 | 61 | 65 | 22.1d |
| 4 | 2,3-Butanedione | 34 | 18 | 70 | 2.5 |
| 5 | Acetone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2,5-hexanedione (10:1) :2,5-hexanedione (10:1) |
61 | 53 | 59 | 3.5 |
| 6 | THF | 36 | 40 | 59 | 8.7 |
| 7 | 1,4-Dioxane | 17 | 6 | 20 | 1.9 |
| 8 | t BuOH | 19 | 0 | — | 1.0 |
| 9 | t BuOH/H2O | 13 | 0 | — | 1.0 |
| 10 | H2O | 6 | — | — | — |
| 11 | DMSO | 44 | 18 | 13 | 1.9 |
| 12 | Acetonitrile | 14 | 1 | 13 | 1.1 |
Despite a broad range of alternative chiral ligands being screened, in our hands, it was only PhPyBox that permitted effective KR. Ligand classes screened that were inferior to PhPyBox included other PyBox variants,28 PhOx ligands,29 Trost ligands,30 phosphoramidites,31 BINAP32 and BINOL.33 For full details of ligand screening see ESI.† Further investigation is needed to probe the ligand requirements for this reaction.
The scope of the benzyl azide was investigated next. Initially we ran experiments using in situ prepared azides (from sodium azide and benzyl halide derivatives). Whilst in situ preparation of azides offers the advantage of minimising the number of discrete azide manipulations,34,35 conversions were very poor. So for this communication we prepared just three more isolated benzyl azides to compare against our benchmark reaction (Table 3, entry 1). We probed increasing steric bulk (Table 3, entry 2) by using 2-phenyl benzyl azide (3b), conversion was essentially unchanged but selectivity factor dropped a little (albeit it a respectable s = 17.5). 4-Methyl benzyl azide (3c) and 3,5-trifluoromethyl benzyl azide (3d) (Table 3, entries 3 and 4 respectively), offered the chance to judge any effects due to electronic parameters. In both cases similar selectivity factors (13.1 and 11.1) were obtained with a slightly lower conversion when 3d was used.
|
|
||||
|---|---|---|---|---|
| Entry | R | Conv.a (%) | e.e. SMb (%) | Selectivity factor (s)c |
| a Conversion determined by inspection of 1H NMR spectra (see ESI). b e.e. of recovered starting material (HPLC). c s = ln[(1 − c)(1 − ee)]/ln[(1 − c)(1 + ee)]. d See Table 2, entry 3. e Average of three s = 17.5 ± 2.0, best unique case s = 19.8. f Average of three s = 13.1 ± 1.7, best unique case s = 14.6. g Average of three s = 11.1 ± 2.8, best unique case s = 14.4. | ||||
| 1 | 2a C6H5 | 46 | 72 | 22.1d |
| 2 | 2b 2-PhC6H4 | 45 | 67 | 17.5e |
| 3 | 2c 4-MeC6H4 | 46 | 65 | 13.1f |
| 4 | 2d 3,5-(CF3)2C6H3 | 39 | 51 | 11.1g |
Finally, as with any kinetic resolution, by judicious choice of reaction time, and therefore conversion, it is possible to obtain higher e.e. of starting material or product. Indeed, in our case we can obtain ≥80% e.e. of 2a (albeit at low conversion), see Scheme 2.
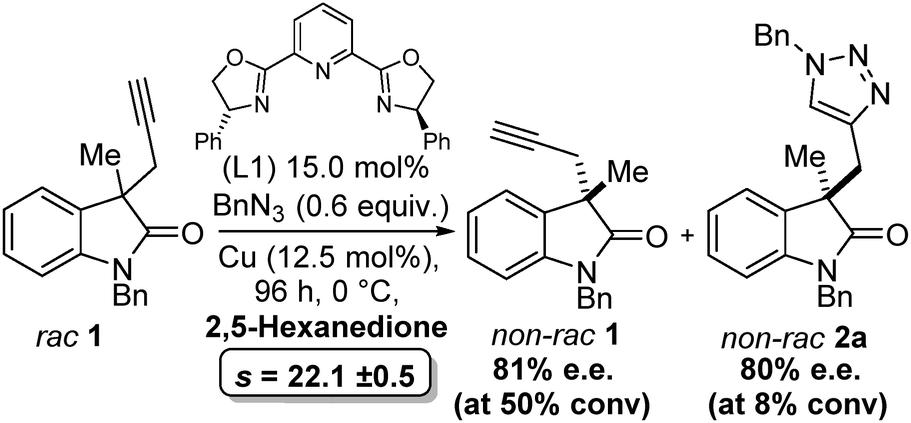 | ||
| Scheme 2 Optimised conditions for the CuAAC kinetic resolution, demonstrating ≥80% e.e. starting material and product can be obtained. | ||
These preliminary findings demonstrate that not only is it possible to perform catalytic kinetic resolution with the CuAAC reaction, but selectivity factors greater than 20 are possible Scheme 2. Optimised conditions for the CuAAC kinetic resolution, demonstrating ≥80% e.e. starting material and product can be obtained with biologically relevant substrates. Order of addition, choice of ligand and choice of solvent were key to achieving reproducible results. It was shown that dependant on reaction conversion high e.e. alkynes are recoverable and these could be used for further derivatisation.
Further expansion of substrate scope and detailed mechanistic studies remain to be carried out and we look forward to the opportunity to report on that in due course.
JSF and WDGB would like to thank the University of Birmingham for support. BRB thanks Loughborough University and Research Councils UK for a RCUK Fellowship. Dr Neil Spencer and Dr Chi Tsang are thanked for helpful discussions about NMR spectroscopy and HPLC analysis respectively. WDGB would also like to thank the RSC and the School of Chemistry at the University of Birmingham for travel support facilitating useful discussions via the CASE Network.36 All investigators are grateful for a Royal Society Research Grant (2012/R1) that underpins this project.
Notes and references
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) W. G. Lewis, L. G. Green, F. Grynszpan, Z. Radić, P. R. Carlier, P. Taylor, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 1053–1057 CrossRef CAS.
- (a) F. Zhou, C. Tan, J. Tang, Y.-Y. Zhang, W.-M. Gao, H.-H. Wu, Y.-H. Yu and J. Zhou, J. Am. Chem. Soc., 2013, 135, 10994–10997 CrossRef CAS; (b) T. Osako and Y. Uozumi, Org. Lett., 2014, 16, 5866–5869 CrossRef CAS; (c) T. Song, L. Li, W. Zhou, Z.-J. Zheng, Y. Deng, Z. Xu and L.-W. Xu, Chem. – Eur. J., 2015, 21, 554–558 CrossRef CAS; (d) G. R. Stephenson, J. P. Buttress, D. Deschamps, M. Lancelot, J. P. Martin, A. I. G. Sheldon, C. Alayrac, A.-C. Gaumont and P. C. B. Page, Synlett, 2013, 2723–2729 CrossRef CAS; (e) Z.-J. Zheng, F. Ye, L.-S. Zheng, K.-F. Yang, G.-Q. Lai and L.-W. Xu, Chem. – Eur. J., 2012, 18, 14094–14099 CrossRef CAS.
- (a) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS; (b) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- (a) H. B. Kagan and J. C. Fiaud, Topics in Stereochemistry, John Wiley & Sons, Inc., 2007, ch. 4, pp. 249–330, DOI: http://10.1002/9780470147276 Search PubMed; (b) J. M. Goodman, A.-K. Köhler and S. C. M. Alderton, Tetrahedron Lett., 1999, 40, 8715–8718 CrossRef CAS.
- (a) K. Tanaka and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 8078–8079 CrossRef CAS; (b) B. Tao, J. C. Ruble, D. A. Hoic and G. C. Fu, J. Am. Chem. Soc., 1999, 121, 5091–5092 CrossRef CAS; (c) J. C. Ruble, H. A. Latham and G. C. Fu, J. Am. Chem. Soc., 1997, 119, 1492–1493 CrossRef CAS; (d) G. C. Fu, Acc. Chem. Res., 2000, 33, 412–420 CrossRef CAS; (e) G. C. Fu, Acc. Chem. Res., 2004, 37, 542–547 CrossRef CAS.
- (a) X. Li, P. Liu, K. N. Houk and V. B. Birman, J. Am. Chem. Soc., 2008, 130, 13836–13837 CrossRef CAS; (b) B. Hu, M. Meng, Z. Wang, W. Du, J. S. Fossey, X. Hu and W.-P. Deng, J. Am. Chem. Soc., 2010, 132, 17041–17044 CrossRef CAS; (c) S. J. Miller, G. T. Copeland, N. Papaioannou, T. E. Horstmann and E. M. Ruel, J. Am. Chem. Soc., 1998, 120, 1629–1630 CrossRef CAS; (d) D. Belmessieri, C. Joannesse, P. A. Woods, C. MacGregor, C. Jones, C. D. Campbell, C. P. Johnston, N. Duguet, C. Concellon, R. A. Bragg and A. D. Smith, Org. Biomol. Chem., 2011, 9, 559–570 RSC; (e) B. Hu, M. Meng, J. S. Fossey, W. Mo, X. Hu and W.-P. Deng, Chem. Commun., 2011, 47, 10632–10634 RSC; (f) Z. Wang, J. J. Ye, R. Wu, Y. Z. Liu, J. S. Fossey, J. G. Cheng and W. P. Deng, Catal. Sci. Technol., 2014, 4, 1909–1913 RSC.
- J.-c. Meng, V. V. Fokin and M. G. Finn, Tetrahedron Lett., 2005, 46, 4543–4546 CrossRef CAS.
- (a) H. Nishiyama, M. Kondo, T. Nakamura and K. Itoh, Organometallics, 1991, 10, 500–508 CrossRef CAS; (b) D. A. Evans, J. A. Murry, P. Vonmatt, R. D. Norcross and S. J. Miller, Angew. Chem., Int. Ed., 1995, 34, 798–800 CrossRef CAS.
- Selecitivty factor (s) = ln[(1 − c)(1 − ee)]/ln[(1 − c)(1 + ee)] where c = conversion and the ee refers to that of the starting material.
- M. K. Christensen, K. D. Erichsen, C. Trojel-Hansen, J. Tjornelund, S. J. Nielsen, K. Frydenvang, T. N. Johansen, B. Nielsen, M. Schested, P. B. Jensen, M. Ikaunieks, A. Zaichenko, E. Loza, I. Kalvinsh and F. Bjorkling, J. Med. Chem., 2010, 53, 7140–7145 CrossRef CAS.
- H. B. Rasmussen and J. K. MacLeod, J. Nat. Prod., 1997, 60, 1152–1154 CrossRef CAS.
- K. Monde, K. Sasaki, A. Shirata and M. Takasugi, Phytochemistry, 1991, 30, 2915–2917 CrossRef CAS.
- K. Tchabanenko, N. S. Simpkins and L. Male, Org. Lett., 2008, 10, 4747–4750 CrossRef CAS.
- M. Panera, J. Díez, I. Merino, E. Rubio and M. P. Gamasa, Inorg. Chem., 2009, 48, 11147–11160 CrossRef CAS.
- (a) M. Ahlquist and V. V. Fokin, Organometallics, 2007, 26, 4389–4391 CrossRef CAS; (b) B. R. Buckley, S. E. Dann, D. P. Harris, H. Heaney and E. C. Stubbs, Chem. Commun., 2010, 46, 2274–2276 RSC; (c) B. R. Buckley, S. E. Dann and H. Heaney, Chem. – Eur. J., 2010, 16, 6278–6284 CrossRef CAS; (d) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS.
- G.-C. Kuang, P. M. Guha, W. S. Brotherton, J. T. Simmons, L. A. Stankee, B. T. Nguyen, R. J. Clark and L. Zhu, J. Am. Chem. Soc., 2011, 133, 13984–14001 CrossRef CAS.
- (a) L. Jin, D. R. Tolentino, M. Melaimi and G. Bertrand, Sci. Adv., 2015, 1 DOI:10.1126/sciadv.1500304; (b) A. Makarem, R. Berg, F. Rominger and B. F. Straub, Angew. Chem., Int. Ed., 2015, 54, 7431–7435 CrossRef CAS.
- B. R. Buckley and D. R. B. Fernandez, Tetrahedron Lett., 2013, 54, 843–846 CrossRef CAS.
- C. Abbadie, O. B. McManus, S.-Y. Sun, R. M. Bugianesi, G. Dai, R. J. Haedo, J. B. Herrington, G. J. Kaczorowski, M. M. Smith, A. M. Swensen, V. A. Warren, B. Williams, S. P. Arneric, C. Eduljee, T. P. Snutch, E. W. Tringham, N. Jochnowitz, A. Liang, D. Euan MacIntyre, E. McGowan, S. Mistry, V. V. White, S. B. Hoyt, C. London, K. A. Lyons, P. B. Bunting, S. Volksdorf and J. L. Duffy, J. Pharmacol. Exp. Ther., 2010, 334, 545–555 CrossRef CAS.
- A. D. Laird, P. Vajkoczy, L. K. Shawver, A. Thurnher, C. Liang, M. Mohammadi, J. Schlessinger, A. Ullrich, S. R. Hubbard, R. A. Blake, T. A. T. Fong, L. M. Strawn, L. Sun, C. Tang, R. Hawtin, F. Tang, N. Shenoy, K. P. Hirth, G. McMahon and J. M. Cherrington, Cancer Res., 2000, 60, 4152–4160 CAS.
- (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS; (b) Y. Kamano, H.-p. Zhang, Y. Ichihara, H. Kizu, K. Komiyama and G. R. Pettit, Tetrahedron Lett., 1995, 36, 2783–2784 CrossRef CAS; (c) A. Natarajan, Y. Guo, F. Harbinski, Y.-H. Fan, H. Chen, L. Luus, J. Diercks, H. Aktas, M. Chorev and J. A. Halperin, J. Med. Chem., 2004, 47, 4979–4982 CrossRef CAS.
- (a) M. M. M. Santos, Tetrahedron, 2014, 70, 9735–9757 CrossRef CAS; (b) T. Tokunaga, W. E. Hume, T. Umezome, K. Okazaki, Y. Ueki, K. Kumagai, S. Hourai, J. Nagamine, H. Seki, M. Taiji, H. Noguchi and R. Nagata, J. Med. Chem., 2001, 44, 4641–4649 CrossRef CAS.
- (a) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS; (b) D. Tomita, K. Yamatsugu, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 6946–6948 CrossRef CAS; (c) J.-S. Yu, F.-M. Liao, W.-M. Gao, K. Liao, R.-L. Zuo and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 7381–7385 CrossRef CAS.
- B. M. Trost, D. A. Bringley, T. Zhang and N. Cramer, J. Am. Chem. Soc., 2013, 135, 16720–16735 CrossRef CAS.
- C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef CAS.
- (a) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS; (b) A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 2794–2819 CrossRef; (c) Y. Jiang, Y. Wei, X.-Y. Tang and M. Shi, Chem. – Eur. J., 2015, 21, 7675–7681 CrossRef CAS.
- Personal communication J. Zhou.
- (a) G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2003, 103, 3119–3154 CrossRef CAS; (b) H. Nishiyama, H. Sakaguchi, T. Nakamura, M. Horihata, M. Kondo and K. Itoh, Organometallics, 1989, 8, 846–848 CrossRef CAS.
- (a) G. J. Dawson, C. G. Frost and J. M. J. Williams, Tetrahedron Lett., 1993, 34, 3149–3150 CrossRef CAS; (b) J. Sprinz and G. Helmchen, Tetrahedron Lett., 1993, 34, 1769–1772 CrossRef CAS; (c) P. von Matt and A. Pfaltz, Angew. Chem., Int. Ed., 1993, 32, 566–568 Search PubMed.
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS.
- J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486–2528 CrossRef CAS.
- R. Noyori and H. Takaya, Acc. Chem. Res., 1990, 23, 345–350 CrossRef CAS.
- J. M. Brunel, Chem. Rev., 2007, 107, PR1–PR45 CrossRef CAS.
- Isolation of organic azides is potentially dangerous. Organic azides in pure forms especially with low molecular weights or C:N ratios <2 can spontaneously explode. Extreme care should always be used when handling and isolating organic azides.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef.
- J. S. Fossey and W. D. G. Brittain, Org. Chem. Front., 2015, 2, 101–105 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, HPLC traces, NMR data and detailed screening tables. See DOI: 10.1039/c5cc04886a |
| ‡ The project was conceived equally by JSF and BRB. Experiments were performed at the University of Birmingham hence JSF is corresponding author for experimental details, but both BRB and JSF may be contacted about this manuscript. |
| This journal is © The Royal Society of Chemistry 2015 |