
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The mechanism of CO2 hydration: a porous metal oxide nanocapsule catalyst can mimic the biological carbonic anhydrase role†‡
Nuno A. G.
Bandeira
a,
Somenath
Garai
b,
Achim
Müller
b and
Carles
Bo
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), Avda. Països Catalans, 16. 43007 Tarragona, Spain. E-mail: cbo@iciq.cat
bFakultät für Chemie, Universität Bielefeld, Postfach 100131, 33501 Bielefeld, Germany
cDepartment Química Física i Inorgànica, University Rovira i Virgili, Marcel li Domingo s/n, 43007 Tarragona, Spain
First published on 28th August 2015
Abstract
The mechanism for the hydration of CO2 within a Keplerate nanocapsule is presented. A network of hydrogen bonds across the water layers in the first metal coordination sphere facilitates the proton abstraction and nucleophilic addition of water. The highly acidic properties of the polyoxometalate cluster are crucial for explaining the catalysed hydration.
Concerns about global warming, together with the incoming necessity to find alternative feedstocks to fossil fuels,1 have boosted interest in the capture and use of CO2 as a chemical starting material.2–5 Living organisms having the carbonic anhydrase enzyme carry out the simplest CO2 transformation, i.e. hydration to carbonic acid, in an easy manner. The presence of an electrophilic Zn center together with a network of water molecules in the proximity of the enzyme site makes the hydration reaction possible, which is rather slow in the absence of a catalyst. The exploration of carbonic anhydrase6–9 and related analogues10 has afforded major bio-inspired catalytic routes for CO2 fixation over the past few decades. On the other hand, synthetic chemistry afforded new transition metal based catalysts that can convert CO2 into other chemical entities, for instance CO2 reduction to methanol,11 coupling with oxiranes to produce cyclic carbonates,12–14 or other value added chemicals.15,16
Some of us reported recently17 a novel way to sequestrate and transform CO2 into carbonate by encapsulation within unique molybdenum oxide nanocapsules. This novelty hinges on the fact that the approach uses only aqueous, room temperature and open air chemistry. These capsules, belonging to the Keplerate family, are nano-sized molecular metal oxide spheres with the general formula [{(MVI)MVI5O21(H2O)6}12{M′V2O2X2(μ2-Y)}30]n− (M = Mo, W; M′ = Mo; X = O, S; Y = bridging ligand, e.g. RCOO−, SO42−).18 This sort of capsule contains 12 pentagonal {MoVI6} units placed at the vertices of an icosahedron and linked by 30 binuclear {MoV2} units. This arrangement leads to the formation of capsules (Fig. 1) with twenty {M3Mo6O9}-type pores and a cavity where a large quantity of water molecules, anions or other species can be confined.19,20 By bubbling CO2 in an aqueous solution of (NH4)42[{(MoVI)MoVI5O21(H2O)6}12{MoV2O4(μ2-CH3COO)}30]·ca. 10 CH3COONH4·ca. 300 H2O ≡ (NH4)42·Anion 1a·ca. 10 CH3COONH4·ca. 300 H2O ≡ Compound 121 at pH 7 the carbonate derivative (NH4)72 [{(MoVI)MoVI5O21(H2O)6}12{MoV2O4(μ2-CO3)}30]·ca. 260 H2O ≡ (NH4)72 anion 2a·ca. 260 H2O ≡ Compound 2 was obtained.17 The pictorial representation of the {Mo132} Keplerate capsule is displayed in Fig. 1. The characterisation of Compound 2 prompted the major question of whether the carbonate anion formed in solution (in minute amounts at pH 7) was captured by the Keplerate cluster by diffusion into the inner cavity or, more interestingly, whether the carbonate anion formation took place in situ inside the capsule, either at the MoV or MoVI coordination sites, by a metal catalysed nucleophilic addition of water to a solubilised CO2 molecule, likewise the accepted mechanism of carbonic anhydrase.
 | ||
| Fig. 1 The pictorial representation of the {Mo132} Keplerate capsule. | ||
The CO2 transformation is also reversible via acidification of the aqueous solution of Compound 2.17 The results of the theoretical study presented herein suggest that this transformation of CO2 to carbonate is actually the third example22,23 known to date of a catalytic process occurring inside the {Mo132} capsule, where the MoV and also the MoVI sites play a role.
The mechanism of the hydration of CO2 to form the carbonic acid has been a subject of theoretical studies over the past few decades.24,25 The challenge lies in the accurate description of the explicit water molecules participating in the reaction as was shown by the latest work of Yamabe and Kawagishi.26 The uppermost energy barrier of carbon dioxide hydration is always the initial step of water addition.27 The arrangement of this initial transition state24–26,28 is a cyclic three water molecular arrangement as depicted in Fig. 2. We will adopt this model as a benchmark to compare with our own calculations on the catalytic sequestration of CO2 and its conversion into the carbonate form.
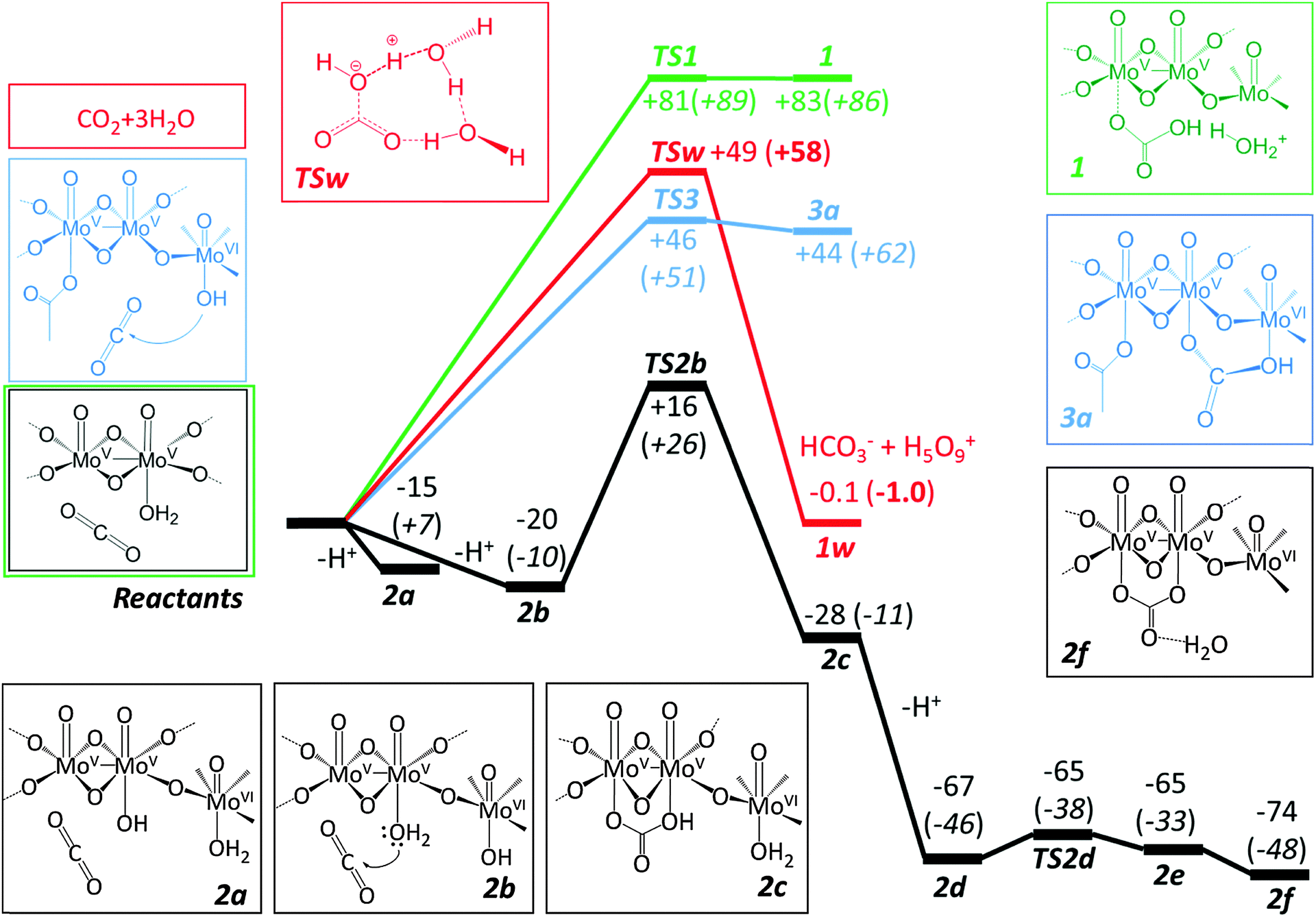 | ||
| Fig. 2 Several mechanistic pathways for CO2 hydration: in red the uncatalysed reaction is presented; in green the iso-charge pathway leading to the formation of a local H3O+ cation; in blue is the route with direct MoVI intervention in the formation of bicarbonate; in black the proposed catalytic pathway. Electronic energies and Gibbs free energies in parentheses are evaluated using a partial Hessian. All energies in kJ mol−1. | ||
In a recent study we demonstrated that by using a cluster model of the {Mo132} nanocapsule, the reaction pathway of the reversible cleavage of methyl-tert-butyl ether22 was successfully unravelled. The model assembly was defined to mimic the nature of the active sites of the Keplerate and it was formulated as [{(MoVI)MoVI5O13(OH)8}2{MoV2O4}]6+ containing two pentagonal {(MoVI)MoVI5}-units and one linker unit of the type {MoV2O4}. It fully retained the essential characteristics of the {Mo132} reactive sites and therefore we have selected that model for the present study. Since the formation of the carbonate anion takes place in aqueous media, the presence of water molecules inside the Keplerate sphere must play an essential role in the reaction and therefore it is essential that the cluster model should incorporate a sufficiently large number of water molecules. Thus we included 13 additional water molecules explicitly in this study, so the model used is formulated as [{(MoVI)MoVI5O13(H2O)6(OH)8}2{MoV2O4(H2O)}]6+, which leaves one vacant coordination site reserved for the incoming CO2 molecule at one of the two MoV sites, while the second one bears a water molecule which is supposed to undergo nucleophilic addition.
As expected the CO2 molecule, being nonpolar, does not coordinate in the initial stage to an MoV centre either in an η1 or η2 fashion. Notwithstanding, we could characterize a weakly bound stationary point structure in which CO2 is hydrogen-bonded to the water molecule in one MoV centre and to a water molecule on MoVI, thus located in the vicinity of the reactive centre. This will be our starting point (named Reactants) for the reaction path studies defining the zero of energies.
The highest energy reaction path explored TS1 (Fig. 2) is perhaps the most intuitive pathway involving a concerted nucleophilic addition of an aqua ligand to CO2 followed by the subsequent proton rejection and formation of a local Zundel cation (H5O2+) sponsored by the hydrogen bonding of the neighbouring aqua ligands. Note that the neighbouring water ligands coordinated to MoVI centres contribute to stabilizing the rejected proton and the concomitant formation of bicarbonate. Although we explored multiple conformational possibilities, a coordinated adduct of the type {O2C–OH2} could not be obtained.
Owing to the accumulation of positive charge closer to the metal centres, TS1 transition state is shown to be too excessively high in energy (+89 kJ mol−1) to become a competitive pathway vis-à-vis the unassisted TSw transition state for hydration of CO2.
In light of these results we explored a different route that yielded a bicarbonate coordinated intermediate resulting from the nucleophilic addition of a hydroxo group to CO2. Given that the MoVI centres are more Lewis acidic than MoV (e.g. on average 0.1 pKa units lower in MoVI for the Keggin structure29) the likely candidate for a good reactant would be 2b bearing the {MoV(OH2)–O–MoVI(OH)} unit rather than 2a ({MoV(OH)–O–MoVI(OH2)}). This is borne out by the relative energetics of the two isomers, which favour 2b by some 5 kJ mol−1. The mechanism should expectedly involve a proton relay from the aqua-ligand in the MoV centre concerted with the nucleophilic addition of the hydroxo group to CO2. The ΔG estimate for the 2a → 2b conversion is further widened to 17 kJ mol−1 in favour of 2b. The reason for this will be discussed below.
An in vacuo fragment analysis shows that the interaction energy in 2b between the CO2 molecule and the metalate cluster is −39.3 kJ mol−1 showing a weak interaction between them.
It should be mentioned that throughout the process the local MoV character of the linker units is retained throughout the process, i.e. the 4d orbital contribution in the HOMO rests mostly in the 4dσ–4dσ interaction localised on the linker unit.
The bicarbonate intermediate undergoes further deprotonation resulting in 2d. The release of a proton from 2d to 2e has a negligible energy barrier (for TS2d, 2 kJ mol−1 in electronic or + 8 kJ mol−1 in free energy). The carbonate intermediate 2e is approximately iso-energetic with its parent bicarbonate 2d but can be easily converted to 2f with lower free energy. The intermediate 2f has one non-coordinated water molecule which stabilises the carbonate ligand via hydrogen bonding. The Mo–carbonate bond lengths in 2e are 2.392 and 2.329 Å, which are within the error limits of the experimentally determined values.12
The higher acidity of the MoVI centre prompted us to explore another possible mechanistic route in which the direct nucleophilic addition to the CO2 molecule takes place directly by the hydroxo group coordinated to the MoVI sites while the vacant coordination site of MoV is utilized to stabilize the transition state. A subsequent backflip of bicarbonate or carbonate to the {MoV2}-linker would be necessary to be consistent with the final carbonate adduct. The initial steps of this pathway are sketched in blue as shown in Fig. 2. The transition state TS3 has a similar energy value to TSw (the uncatalysed transition state) but intermediate 3a is not sufficiently stable to be considered a viable route (see ESI,‡ for these additional structures).
There are structural differences between the catalysed and uncatalysed systems namely with regard to each transition state which are summarised in Fig. 3. The Mayer–Mulliken bond orders30 (MBOs) were also analysed in the present case which reflect the bond strength between the different atoms in any given system. The most striking difference between TSw and TS2b is that the latter is a slightly “lesser bound” transition state with a reaction coordinate (C–O) bond order 0.377 whereas in TSw it is 0.557 in line with Hammond's postulate. The ∠(O–C–O) angles are also considerably different between TSw (139°) and TS2b (152°) consistent with a larger electron cloud of the incoming O(–C) and consequently a lower angular distortion of CO2. The leaving proton is also more bound to the oxygen atom in TSw (MBO = 0.430) than in TS2b (MBO = 0.250). In the latter case the outgoing proton from the aqua ligand is already at a large distance (1.535 Å, see Fig. 3). Interestingly the MoV–OH bond in 2a (α-hydroxo isomer) is stronger (MBO = 0.450) than the MoVI–OH bond in 2b (MBO = 0.219). This causes a vibrational stiffness in the 2a isomer decreasing its entropy and increasing the free energy difference with respect to 2b.
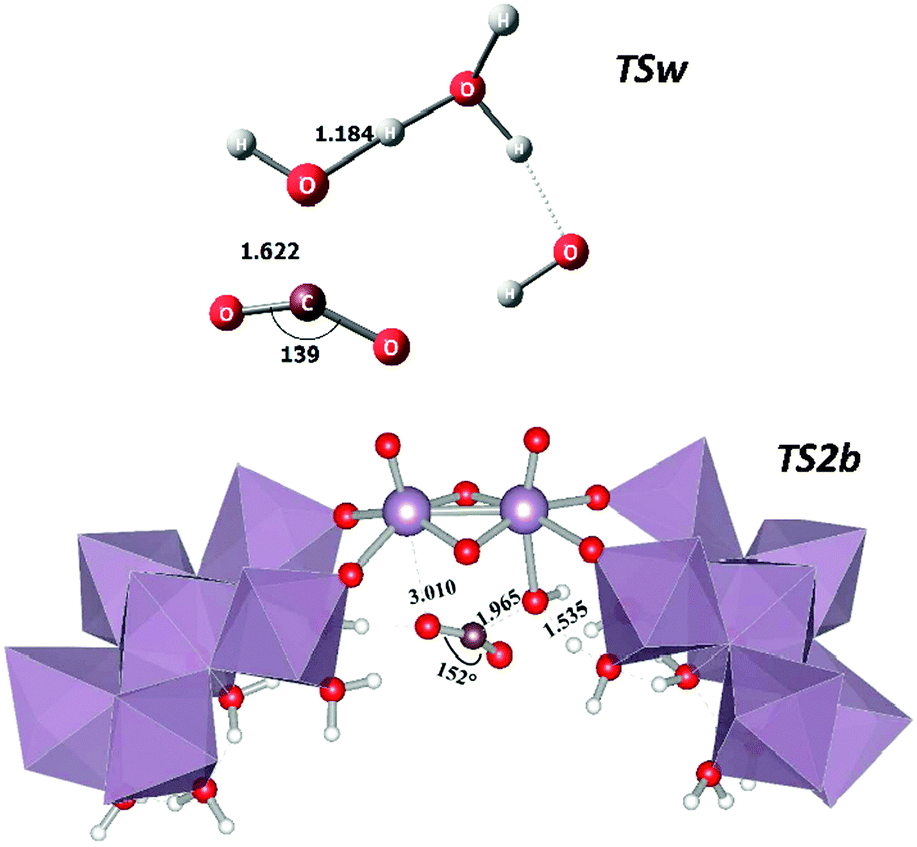 | ||
| Fig. 3 Transition state structures for the uncatalysed CO2 + 3H2O (TSw) and for the catalysed reaction (TS2b). Selected distances in Å and angles in degrees. | ||
Finally to predict the potential reactivity of related systems, additional calculations were carried out on model analogues of the {W72Mo60} and {W132} nanocapsules. The former nanocapsule has been characterised31 experimentally although the latter is still unknown. Since the key point in the mechanism is the generation of the nucleophilic hydroxo species coordinated to the star-shaped MVI moieties, the relative thermodynamic stability of 2a and 2b species was determined. The calculated ΔE(2a → 2b) is −65 kJ mol−1 for the mixed W/Mo oxo-cluster model and −85 kJ mol−1 for the hypothetical full W system. This points to a likely enhanced reactivity of the heavier metal Keplerates in the order {Mo132} < {W72Mo60} < {W132}. These results also indicate that WV centres are less (Lewis) acidic with respect to WVI than MoV in relation to MoVI.
DFT based calculations enabled unravelling the CO2 hydration reaction pathway as evidenced involving Compound 1 by considering the known mechanism in the aqueous solution. The in situ bicarbonate formation, promoted by the MoV centres, inside the capsule is kinetically more favourable than direct carbonate uptake from aqueous solution. Three trials were performed in the present work, which can be summarised as follows:
(i) A neutral charge pathway with aqua ligand nucleophilic addition to CO2 results in a high kinetic barrier ΔE‡ = +81 kJ mol−1 and a product of exceedingly high energy.
(ii) A hydroxo ligand pathway in which the nucleophilic attack takes place on a MoVI site. This is a high energy process requiring +44 kJ mol−1 at the calculation level to form a product.
(iii) A hydroxo ligand pathway where the hydroxo group in an MoVI centre will act as a proton acceptor in tandem with the nucleophilic addition of CO2 to an aqua ligand at the MoV sites. The activation energy ΔE‡ = +36 kJ mol−1 is the lowest of all the trials, even lower than the uncatalysed hydration reaction, and the ensuing product assembly is 28 kJ mol−1 more stable than the reactant assembly.
Therefore the most plausible mechanism for the formation of Compound 2 will be the latter based on comparison of computed energies with respect to a comparable micro-solvated CO2 hydration. The resemblance of the mechanism with that operating in the carbonic anhydrase enzyme is remarkable. The subtle differences lie in the first steps of the latter mechanism: the rate-limiting step is the protolysis of the aqua ligand in (His)3Zn–OH23b,4 which is then followed by a lower energy nucleophilic addition to CO2 whereas the Keplerate acts in a concerted single step for both. These results pave the way for defining a new application of Keplerate anionic capsules as CO2 storage nanodevices.
While the MoV sites still remain the active catalytic host in Keplerate catalysis there is a clear involvement of the adjacent MoVI centres as promoters of Brønsted acidity and proton relay.
This work was funded by the Spanish Ministerio de Economia y Competitividad (MINECO) through project CTQ2014-52824-R, by the Generalitat de Catalunya project 2014SGR409, and by the ICIQ Foundation. The Severo Ochoa Excellence Accreditation (SEV-2013-0319) and the COST Action CM1203 “Polyoxometalate Chemistry for Molecular Nanoscience (PoCheMoN)” are gratefully acknowledged. NAGB gratefully acknowledges COFUND/Marie Curie action 291787-ICIQ-IPMP for funding. A.M. acknowledges continuous financial support by the Deutsche Forschungsgemeinschaft and the ERC (Brussels) for an Advanced Grant.
Notes and references
- M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley, 2010 Search PubMed.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 CAS.
- J. J. Vericella, S. E. Baker, J. K. Stolaroff, E. B. Duoss, J. O. Hardin IV, J. Lewicki, E. Glogowski, W. C. Floyd, C. A. Valdez, W. L. Smith, J. H. Satcher Jr, W. L. Bourcier, C. M. Spadaccini, J. A. Lewis and R. D. Aines, Nat. Commun., 2015, 6, 6124 CrossRef CAS PubMed.
- V. Bhola, F. Swalaha, R. Ranjith Kumar, M. Singh and F. Bux, Int. J. Environ. Sci. Technol., 2014, 11, 2103–2118 CrossRef CAS.
- A. Sanna, M. Uibu, G. Caramanna, R. Kuusik and M. M. Maroto-Valer, Chem. Soc. Rev., 2014, 43, 8049–8080 RSC.
- K. D’Ambrosio, G. De Simone and C. T. Supuran, in Carbonic Anhydrases as Biocatalysts, ed. G. De Simone and C. T. Supuran, Elsevier, Amsterdam, 2015, pp. 17–30 Search PubMed.
- D. N. Silverman and R. McKenna, Acc. Chem. Res., 2007, 40, 669–675 CrossRef CAS PubMed.
- C. Greco, V. Fourmond, C. Baffert, P.-h. Wang, S. Dementin, P. Bertrand, M. Bruschi, J. Blumberger, L. de Gioia and C. Leger, Energy Environ. Sci., 2014, 7, 3543–3573 CAS.
- J. K. J. Yong, G. W. Stevens, F. Caruso and S. E. Kentish, J. Chem. Technol. Biotechnol., 2015, 90, 3–10 CrossRef CAS PubMed.
- G. Parkin, Chem. Rev., 2004, 104, 699–768 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- K. R. Roshan, B. M. Kim, A. C. Kathalikkattil, J. Tharun, Y. S. Won and D. W. Park, Chem. Commun., 2014, 50, 13664–13667 RSC.
- F. Castro-Gómez, G. Salassa, A. W. Kleij and C. Bo, Chem. – Eur. J., 2013, 19, 6289–6298 CrossRef PubMed.
- C. J. Whiteoak, A. H. Henseler, C. Ayats, A. W. Kleij and M. A. Pericàs, Green Chem., 2014, 16, 1552–1559 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef CAS PubMed.
- A. T. Najafabadi, Renewable Sustainable Energy Rev., 2015, 41, 1515–1545 CrossRef PubMed.
- S. Garai, E. T. K. Haupt, H. Bögge, A. Merca and A. Müller, Angew. Chem., Int. Ed., 2012, 51, 10528–10531 CrossRef CAS PubMed.
- A. Müller and P. Gouzerh, Chem. Soc. Rev., 2012, 41, 7431–7463 RSC.
- T. Mitra, P. Miró, A.-R. Tomsa, A. Merca, H. Bögge, J. B. Ávalos, J. M. Poblet, C. Bo and A. Müller, Chem. – Eur. J., 2009, 15, 1844–1852 CrossRef CAS PubMed.
- C. Schäffer, A. M. Todea, H. Bögge, O. A. Petina, D. Rehder, E. T. K. Haupt and A. Müller, Chem. – Eur. J., 2011, 17, 9634–9639 CrossRef PubMed.
- A. Müller, E. Krickemeyer, H. Bögge, M. Schmidtmann and F. Peters, Angew. Chem., Int. Ed., 1998, 37, 3360–3363 Search PubMed.
- S. Kopilevich, A. Gil, M. Garcia-Ratés, J. Bonet-Ávalos, C. Bo, A. Müller and I. A. Weinstock, J. Am. Chem. Soc., 2012, 134, 13082–13088 CrossRef CAS PubMed.
- C. Besson, S. Schmitz, K. M. Capella, S. Kopilevich, I. A. Weinstock and P. Kögerler, Dalton Trans., 2012, 41, 9852–9854 RSC.
- C. S. Tautermann, A. F. Voegele, T. Loerting, I. Kohl, A. Hallbrucker, E. Mayer and K. R. Liedl, Chem. – Eur. J., 2002, 8, 66–73 CrossRef CAS.
- M. T. Nguyen, M. H. Matus, V. E. Jackson, V. T. Ngan, J. R. Rustad and D. A. Dixon, J. Phys. Chem. A, 2008, 112, 10386–10398 CrossRef CAS PubMed.
- S. Yamabe and N. Kawagishi, Theor. Chem. Acc., 2011, 130, 909–918 CrossRef CAS.
- M. J. Welch, J. F. Lifton and J. A. Seck, J. Phys. Chem., 1969, 73, 3351–3356 CrossRef CAS.
- G. A. Gallet, F. Pietrucci and W. Andreoni, J. Chem. Theory Comput., 2012, 8, 4029–4039 CrossRef CAS.
- G. M. Maksimov, Russ. Chem. Rev., 1995, 64, 445 CrossRef PubMed.
- I. Mayer, Int. J. Quantum Chem., 1986, 29, 73–84 CrossRef CAS PubMed.
- C. Schäffer, A. Merca, H. Bögge, A. M. Todea, M. L. Kistler, T. Liu, R. Thouvenot, P. Gouzerh and A. Müller, Angew. Chem., Int. Ed., 2009, 48, 149–153 CrossRef PubMed.
Footnotes |
| † Dedicated to the memory of Tom Ziegler (1945–2015). |
| ‡ Electronic supplementary information (ESI) available: Computational details and methodology. See DOI: 10.1039/c5cc06423f |
| This journal is © The Royal Society of Chemistry 2015 |