
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence5,5′-Diamino-BIPHEP ligands bearing small selector units for non-covalent binding of chiral analytes in solution†
G.
Storch
,
M.
Siebert
,
F.
Rominger
and
O.
Trapp
*
Organisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany. E-mail: trapp@oci.uni-heidelberg.de
First published on 4th September 2015
Abstract
A dynamic axially chiral BIPHEP-ligand with 3,5-dichlorobenzoyl amide selector units for non-covalent binding of phenylalanine derivatives has been developed. Interaction studies in solution were performed with rhodium(I) complexes under exclusion of the metal being involved in binding. (Rax, SPhe) and (Sax, SPhe) adducts were observed as significantly separated species in NMR spectroscopy.
Interactions between chiral substances represent a fundamental principle in chiral amplification, self-organisation and chirality sensing. This requires covalent, coordinative or non-covalent binding of the chiral molecules. Impressive examples of enantioselective self-assembly1 and binding of chiral compounds2 have been reported in supramolecular chemistry for capsules and clusters.
On a molecular level, it has been shown that metal complexes are able to differentiate between enantiomers in a quite effective way. One interesting field of application is the sensing of enantiomeric excesses of small chiral molecules by complexation to transition metals bearing chiral ligands. Canary et al.3,4 and Anslyn et al.3,5 reported copper complexes with chiral ligands for direct enantiomeric excess determination via CD spectroscopy or colorimetric analysis. Wolf et al.6 demonstrated palladium complexes with dynamic chiral ligands for this purpose.
The formation of diastereomeric substrate–catalyst adducts that occur in metal catalyzed enantioselective hydrogenation (re or si coordination) represents a related case of asymmetric recognition.7 The vice versa process of metal-coordinated and (−)-menthol functionalised substrates, selectively coordinating one additional phosphine ligand enantiomer upon addition of a racemic ligand mixture, has been shown as well.8
Impressive results in enantioselective catalysis have been shown by Mikami et al. utilizing chiral diamines as aligning ligands for the control of a dynamic chiral phosphine ligand.9,10 Previously, Mikami and Noyori reported ruthenium and titanium complexes bearing racemic ligands in combination with enantiopure diamine or biphenol co-ligands as efficient catalytic systems for asymmetric activation.11,12 Faller et al. used a similar approach with racemic rhodium complexes and chiral phosphorous ligand additives.13,14
Since free coordination sites are blocked by the aligning ligands, they often have to be cleaved off prior to catalysis.15 Brown et al. and Mikami et al. reported a strategy to overcome this limitation by using elaborated enantiopure chiral COD-analogues that are cleaved off under catalysis conditions for alignment of dynamic chiral ligands.16,17 Another approach focused on the use of chiral ionic liquids with free amino functionalities for metal coordination.18,19
However, the common ground of all these findings is that a chiral analyte is introduced via metal coordination. Not only does this render the metal centre a prerequisite for the concept to work; it also often limits further catalytic applications at the metal centre. Furthermore, all mentioned examples share the requirement of the analyte to bind to the specific metal, e.g. via free amines or phosphines, which intrinsically excludes analytes without these groups. Our investigations were focused on changing this principle and introducing selector groups for non-covalent analyte binding. We consider this new concept as highly promising, because no metal vacancy is blocked by coordination and thus can be utilised for further purposes (Fig. 1).
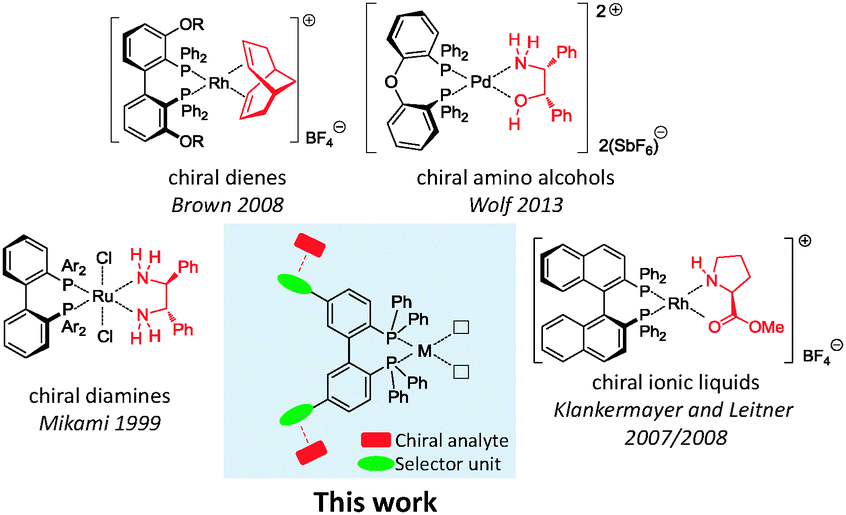 | ||
| Fig. 1 Previous investigations of complexes bearing chiral or dynamic chiral ligands in binding of chiral analytes. The fundamental concept of our new strategy is depicted centred. | ||
We focused on the synthesis of a 5,5′-diamino functionalised BIPHEP ligand enabling selector bonding. Although axially chiral BINAP ligands bearing methylamino20 or amino21 groups in the backbone are known in literature, these molecules were not suitable for our purpose for several reasons: methylamino groups were considered to be too flexible, thus direct bonding of the NH2-moiety was considered to be a more promising approach because the enhanced rigidity should have a beneficial effect on analyte binding. Since future studies will aim at controlling the axial stereochemistry, the tropos nature of BIPHEP-type ligands was also advantageous. Modification of BIPHEP-ligands in the 6-position leads to atropos ligands as it is the case for the widely used MeO-BIPHEP ligand family. In contrast, the 3-position directs the selector to close proximity of the diphenylphosphino groups potentially making substrate coordination difficult due to additional steric repulsion. Little is known about effects of substitution at the 4-position,22 however, it can be assumed that substituents in this position will have less influence on the axial stereochemistry of the BIPHEP-ligand. Therefore, we decided to develop a synthetic strategy for the 5-substituted derivative (Fig. 2) in line with our previously investigated hydroxy functionalised BIPHEPs.23
 | ||
| Fig. 2 Synthetic overview of selector modified BIPHEP oxides. (A) Preparation of the ligand backbone: (i) HOAc, HNO3, 16%, (ii) Tf2O, pyridine, 85%, (iii) Fe, aq. HCl, 84%, (iv) Boc2O, 84%, (v) Pd2(dba)3, dppb, DIPEA, HP(O)Ph2, 26%, (vi) HCl in 2-propanol, 93%; (B) modification of 2 with various types of selectors: (vii) 3,5-dichlorobenzoyl chloride, pyridine, 92%, (viii) N-3,5-dichlorobenzoyl glycine, EEDQ, 60%, (ix) 3,5-dichlorophenyl isocyanate, 52%. | ||
The synthetic procedure starts with nitration of commercially available ortho-biphenol24 and selective crystallisation of the 5,5′-dinitro derivative. Conversion of the phenol groups into triflates and reduction of the nitro groups yielded 1. N-Boc-protection allowed for subsequent palladium catalysed C–P-coupling with HP(O)Ph2. Acidic cleavage thus led to 2 which can easily be isolated as hydrogen chloride salt (Fig. 2A).
Several types of selector units can be attached to ligand backbone 2. We chose state of the art chiral HPLC column selectors, i.e. interacting sites of amylose and cellulose CSPs (Chiralpak IC/IE series) as model and focused on the 3,5-dichlorophenyl moiety attached to the ligand core via carboxylic acids or isocyanates. The resulting amide or urea NH and CO functionalities as well as the electron deficient aromatic rings are known for efficient binding of analytes in HPLC stationary phase design. Reaction with acyl chlorides readily yielded the corresponding amides exemplified by the preparation of 3. Amide bond formation can also be applied to the established coupling of diamides with EEDQ as it was shown for glycine derivative 4. In even closer analogy to HPLC stationary phases, reaction with isocyanates resulted in urea derivative 5 (Fig. 2B).
Selective reduction of the phosphine oxides was achieved with PhSiH3. Subsequent coordination to the metal precursor [Rh(COD)2](BF4) led to the corresponding rhodium(I) complex (Fig. 3A). X-ray crystallographic investigation25 illustrates the twisted biphenyl core with metal coordinated diphenylphosphino groups on one and the rigid 3,5-dichlorobenzoyl amides on the other side (Fig. 3B). Reduction and coordination was investigated for selector modified BIPHEP oxides 3 and 4. However, it was found that the reduction product of 4 gives rise to very broad signals in the NMR-spectra in non-polar solvents rendering interaction studies almost impossible. Since very sharp signals were observed in case of complex 6, we focused on this derivative in the current study.
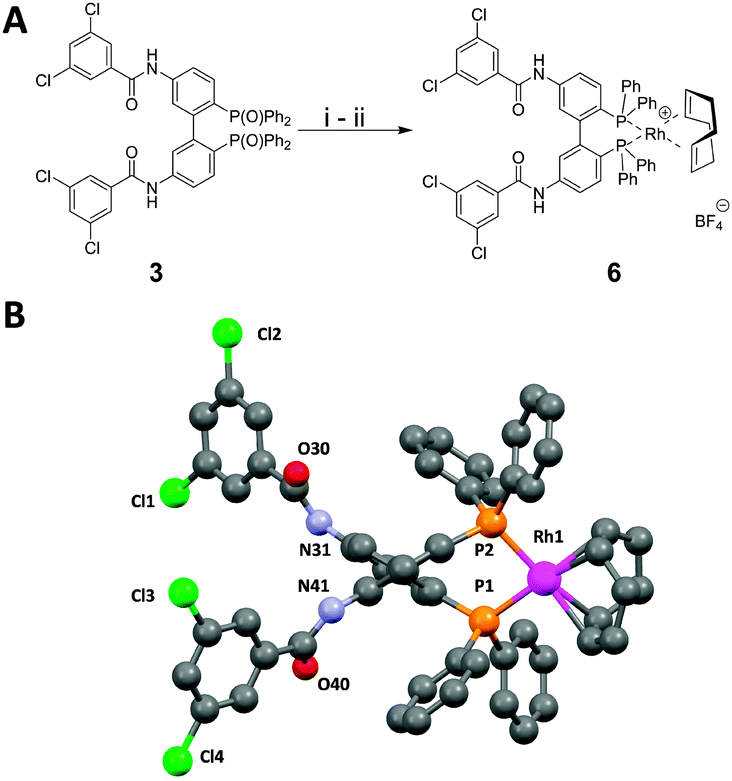 | ||
| Fig. 3 Synthesis and characterization of rhodium(I)-complex 6. (A) Reduction of phosphine oxide 3 and subsequent coordination to a rhodium(I)-precursor: (i) PhSiH3, 70%; (ii) [Rh(COD)2](BF4), 88%; (B) X-ray crystallographic investigation of 6. The depicted structure represents the view along the central biphenyl C–C bond. Hydrogen atoms and the BF4− counter anion are omitted for clarity. | ||
We subsequently investigated derivatives of the amino acid phenylalanine as interacting small chiral analytes. Derivatives of phenylalanine are of great pharmaceutical importance, e.g. in the case of Parkinson's drug L-3,4-dihydroxyphenylalanine (L-DOPA). Besides, they represent the product of enantioselective catalyses, thus samples of varying enantiomeric excess are often analysed. Phenylalanine bears good hydrogen bond donor and acceptor properties and derivatives can easily be obtained by modification of the amino and carboxy functionalities.
Complex 6 was dissolved in CDCl3 and NMR spectra were recorded with two, five and ten equivalents of (S)-phenylalanine (SPhe) derivative. We were interested in signal splitting due to dynamic formation of (Rax, SPhe) and (Sax, SPhe) adducts. (S)-Acetylphenylalanine methyl ester 7 was the first derivative to be investigated. While no interaction was detected with two or five equivalents of substrate, a small splitting (9 ppb) of the complex's NH-groups in 1H-NMR was observed in the case of ten equivalents amidoester (Table 1).
|
|
2 eq.a Δppb 1H (NH), [31P] | 5 eq.a Δppb 1H (NH), [31P] | 10 eq.a Δppb 1H (NH), [31P] | |
|---|---|---|---|---|
| a 6.7 mM 6 in dry degassed CDCl3. Diamide concentration was varied. b Substrate dissolves completely upon warming. c Incomplete dissolution of substrate. | ||||
| 7 |

|
—, [—] | —, [—] | 9, [—] |
| 8 |
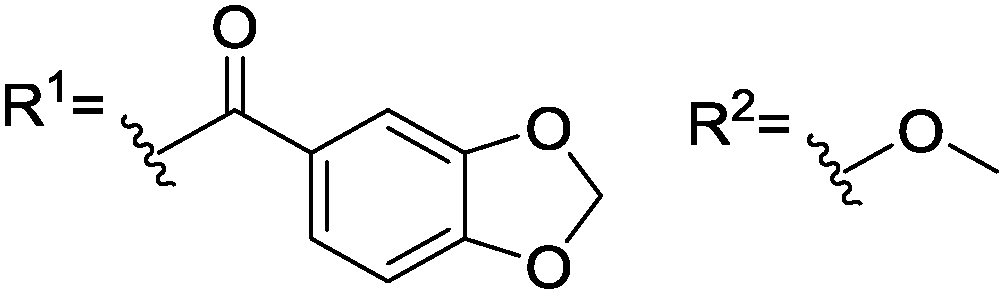
|
—, [—] | —, [—] | —, [—] |
| 9 |

|
92, [—]b | 146, [—]b | 171, [—]c |
| 10 |
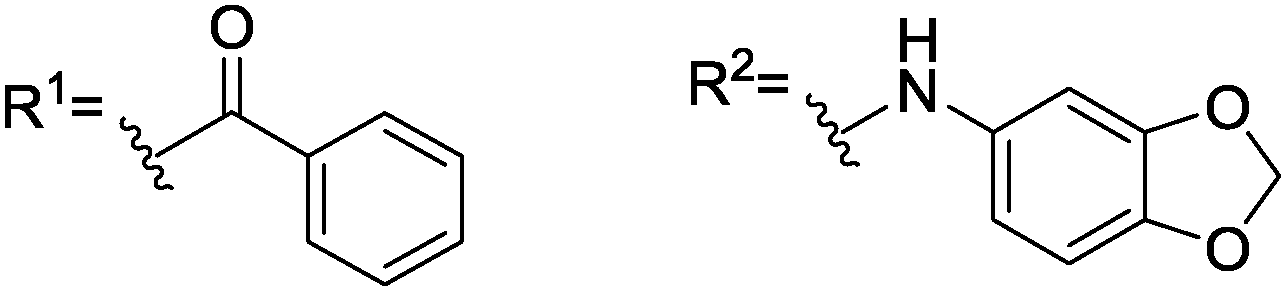
|
—, [12] | 9, [25]b | 22, [37]c |
| 11 |
|
10, [12]b | 27, [25]c | 26, [26]c |
| 12 |

|
57, [27] | 107, [49] | 117, [51]c |
| 13 |

|
322, [10] | 406, [18] | 445, [27]b |
With this result, we implemented electron rich aromatic substituents to the amino acid in order to potentially improve substrate binding efficiency. We thus studied amide bond formation products from piperonyloyl chloride (8) and 3,4-(dioxymethylene)aniline (9). Interestingly, no interaction took place in case of 8 while a splitting of 92 ppb (1H-NMR, amide NH of 6) was already observed in the presence of two equivalents of 9.
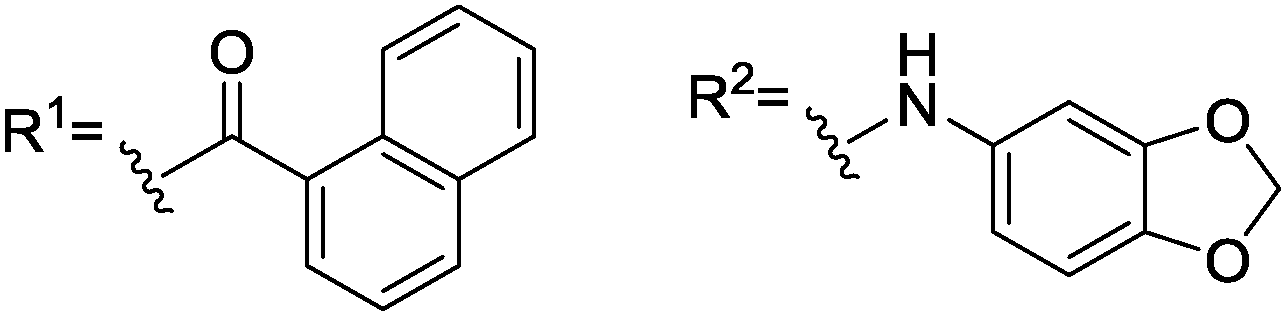
Next, we aimed at increasing the steric demand at the amino group of 9 by replacing the acetyl moiety by benzoyl (10) and 1-naphthoyl (11) groups. These enlarged derivatives should potentially lead to increasing repulsion with the diphenylphosphino groups. Although 10 and 11 were only moderately soluble in CDCl3 (saturated solutions were obtained with less than 10 equivalents), both showed splitting of the 1JP–Rh doublet in 31P{1H}-NMR (Table 1) for the first time.
At this point, we concluded that the most crucial part for strong non-covalent binding would be the presence of two amide bonds. It seemed reasonable to include the methylamide analogues of 7 and 8. Both can easily be obtained by aminolysis of the corresponding methyl esters. N-Piperonyloyl derivative 12 was found to lead to significant splitting in 1H-NMR and remarkable splitting of 0.05 ppm in 31P{1H}-NMR. Intriguingly, the small acetyl derivative 13 gives the highest values regarding splitting in 1H-NMR. The two adducts of (Rax, SPhe) and (Sax, SPhe) are separated by 0.45 ppm (NH signal) in 1H-NMR spectra with 10 equivalents of diamide (Table 1 and Fig. 4A).
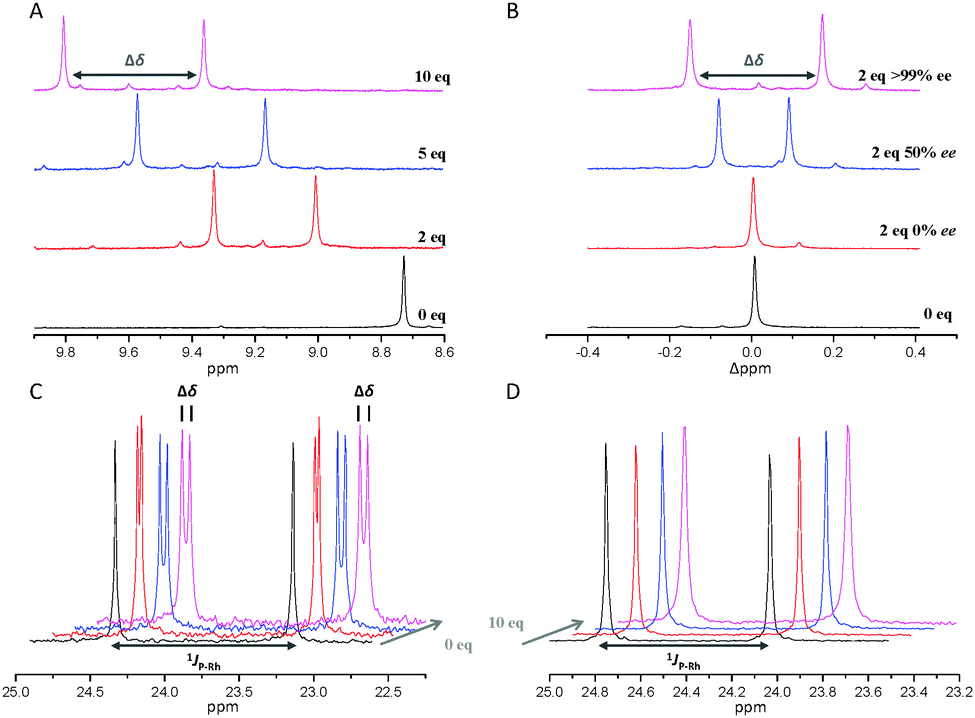 | ||
| Fig. 4 NMR-spectra with varying amounts of phenylalanine analyte. (A) 1H-NMR spectra of 6 with 0, 2, 5 and 10 equivalents (S)-13. The amide NH region of 6 is depicted. (B) 1H-NMR spectra of 6 with 2 equivalents of 13 of 0, 50 and >99% ee (S-enantiomer). The relative deviation of the initial resonance is depicted. (C) 31P{1H}-NMR of 6 with 0, 2, 5 and 10 equivalents of (S)-12. (D) 31P{1H}-NMR of [(BIPHEP)Rh(COD)](BF4) with 0, 2, 5 and 10 equivalents of (S)-12. | ||
In all cases, the splitting effect is not limited to the NH signal in 1H-NMR and can also be observed, for example, when analysing the 3,5-dichlorobenzoyl HAr signals (see ESI†). This supports our initial idea of amide hydrogen bonding and π-stacking of the 3,5-dichlorobenzoyl fragment being the predominant interaction sites.
It has to be noted that binding of the analyte to the metal centre can be excluded because control experiments with [(BIPHEP)Rh(COD)](BF4) did not show any splitting even with ten equivalents of 12 or 13 (Fig. 4D and ESI†). Besides, adding ten equivalents of 13 to the free ligand results in splitting of signals as well (see ESI†).
Next, we verified that the two species observed correspond to the adducts (Rax, SPhe) and (Sax, SPhe). Thus, we repeated the interaction experiment using two equivalents of racemic 13. This led to observation of one single species in the NMR spectrum without any signal splitting. In contrast, signal splitting was observed when adding 13 with 50% ee but the Δppm value was significantly smaller compared to enantiopure 13 (Fig. 4B). It has to be noted, that the two NH-signals do not correspond to RPhe and SPhe, as it would be the case for common chiral shift reagents, thus their relative intensity does not change with different ee values of the analyte. Splitting is the result of the complex sensing the chirality of the analyte through non-covalent coordination to the selector units forming two pairs of adducts: (Rax, SPhe) and (Sax, SPhe). These adducts are diastereomeric and thus are observed at different chemical shifts in NMR spectra. Coordination and de-coordination can occur rapidly in solution. The observed chemical shift of the Rax-enantiomer in case of an analyte of 50% ee is the weighted average of 75% (Rax, SPhe) and 25% (Rax, RPhe). This is why coalescence occurs for the signal splitting of racemic 13 because the observed chemical shift for Rax is the weighted average of 50% (Rax, SPhe) and 50% (Rax, RPhe) while it is the completely enantiomeric 50% (Sax, SPhe) and 50% (Sax, RPhe) for Sax. The splitting correlates linearly with the ee of the added diamide analyte: 0.45 ppm splitting is observed for ten equivalents enantiopure 13, while a value of 0.23 ppm is observed with the same amount of analyte of 50% ee, which is in good agreement with the theoretically halved value (see ESI† for a plot and measurements with analyte of 25 and 75% ee). This represents a rare example of a racemic complex for the determination of ee values by simply measuring NMR spectra and evaluating signal splitting. The method is related to the pro-chiral solvating-agent (pro-CSA) concept recently reported by Hill and Ariga with achiral porphyrine host molecules.26–28
In conclusion, we described the 5,5′-diamino-BIPHEP ligand as a versatile starting point for the synthesis of selector modified stereodynamic ligands. Our investigations were focused on the corresponding complexes obtained with rhodium(I), yet further applications may arise from immobilization of the ligands or formation of ionic adducts.
The rhodium complex bearing a 3,5-dichlorobenzoyl amide functionalized 5,5′-BIPHEP ligand was studied with regard of non-covalent binding of (S)-phenylalanine derivatives in solution. The binding was followed by NMR spectroscopy and observation of signal splitting due to formation of (Rax, SPhe) and (Sax, SPhe) adducts. The extent of splitting was found to be dependent on the substitution pattern of the phenylalanine analyte and additionally on its concentration. Diamides were plausibly found to bind more efficiently compared to amidoester derivatives. Introduction of electron rich aromatic substituents seemed to be of subaltern effect. The extent of signal splitting due to binding of the analytes was also found to be dependent on its enantiomeric excess. This makes ee determination possible.
Our study represents the first example of non-covalent and non-coordinative binding of chiral analytes with a selector modified transition metal complex in solution. These findings open up the perspective of developing self-recognising stereodynamic systems applicable in catalysis and beyond.
Generous financial support by the European Research Council (ERC) for a Starting Grant (No. 258740, AMPCAT) is gratefully acknowledged. G.S. acknowledges the Fonds der Chemischen Industrie for a Ph.D. fellowship.
Notes and references
- H. Jdrzejewska, M. Wierzbicki, P. Cmoch, K. Rissanen and A. Szumna, Angew. Chem., Int. Ed., 2014, 53, 13760 CrossRef PubMed.
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2004, 126, 3674 CrossRef CAS PubMed.
- L. A. Joyce, M. S. Maynor, J. M. Dragna, G. M. da Cruz, V. M. Lynch, J. W. Canary and E. V. Anslyn, J. Am. Chem. Soc., 2011, 133, 13746 CrossRef CAS PubMed.
- Z. Dai, X. Xu and J. W. Canary, Chirality, 2005, 17, S227 CrossRef CAS PubMed.
- J. F. Folmer-Andersen, V. M. Lynch and E. V. Anslyn, J. Am. Chem. Soc., 2005, 127, 7986 CrossRef CAS PubMed.
- P. Zhang and C. Wolf, Chem. Commun., 2013, 49, 7010 RSC.
- J. M. Brown, Organometallics, 2014, 33, 5912 CrossRef CAS.
- N. W. Alcock, J. M. Brown and P. J. Maddox, J. Chem. Soc., Chem. Commun., 1986, 1532 RSC.
- K. Mikami, T. Korenaga, M. Terada, T. Ohkuma, T. Pham and R. Noyori, Angew. Chem., Int. Ed., 1999, 38, 495 CrossRef CAS.
- K. Mikami, K. Aikawa and T. Korenaga, Org. Lett., 2000, 3, 243 CrossRef.
- T. Ohkuma, H. Doucet, T. Pham, K. Mikami, T. Korenaga, M. Terada and R. Noyori, J. Am. Chem. Soc., 1998, 120, 1086 CrossRef CAS.
- K. Mikami and S. Matsukawa, Nature, 1997, 385, 613 CrossRef CAS PubMed.
- J. W. Faller, M. R. Mazzieri, J. T. Nguyen, J. Parr and M. Tokunaga, Pure Appl. Chem., 1994, 66, 1463 CrossRef CAS.
- J. W. Faller and J. Parr, J. Am. Chem. Soc., 1993, 115, 804 CrossRef CAS.
- K. Mikami, H. Kakuno and K. Aikawa, Angew. Chem., Int. Ed., 2005, 44, 7257 CrossRef CAS PubMed.
- T. Punniyamurthy, M. Mayr, A. S. Dorofeev, C. J. R. Bataille, S. Gosiewska, B. Nguyen, A. R. Cowley and J. M. Brown, Chem. Commun., 2008, 5092 RSC.
- K. Aikawa, Y. Takabayashi, S. Kawauchi and K. Mikami, Chem. Commun., 2008, 5095 RSC.
- M. Schmitkamp, D. Chen, W. Leitner, J. Klankermayer and G. Francio, Chem. Commun., 2007, 4012 RSC.
- D. Chen, M. Schmitkamp, G. Franciò, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2008, 47, 7339 CrossRef CAS PubMed.
- (a) R. ter Halle, B. Colasson, E. Schulz, M. Spagnol and M. Lemaire, Tetrahedron Lett., 2000, 41, 643 CrossRef CAS; (b) M. Berthod, C. Saluzzo, G. Mignani and M. Lemaire, Tetrahedron: Asymmetry, 2004, 15, 639 CrossRef CAS PubMed; (c) M. Berthod, G. Mignani and M. Lemaire, J. Mol. Catal. A: Chem., 2005, 233, 105 CrossRef CAS PubMed.
- (a) T. Okano, H. Kumobayashi, S. Akutagawa, J. Kiji, H. Konishi, K. Fukuyama and Y. Shimano, US Pat., 4 705 895, 1987 Search PubMed; (b) Q.-h. Fan, C.-y. Ren, C.-h. Yeung, W.-h. Hu and A. S. C. Chan, J. Am. Chem. Soc., 1999, 121, 7407 CrossRef CAS; (c) Q.-H. Fan, Y.-M. Chen, X.-M. Chen, D.-Z. Jiang, F. Xi and A. S. C. Chan, Chem. Commun., 2000, 789 RSC.
- K. Aikawa, Y. Miyazaki and K. Mikami, Bull. Chem. Soc. Jpn., 2012, 85, 201 CrossRef CAS.
- (a) F. Maier and O. Trapp, Angew. Chem., Int. Ed., 2014, 53, 8756 CrossRef CAS PubMed; (b) F. Maier and O. Trapp, Chirality, 2013, 25, 126 CrossRef CAS PubMed; (c) G. Storch and O. Trapp, Angew. Chem., Int. Ed., 2015, 54, 3580 CrossRef CAS PubMed.
- O. Diels and A. Bibergeil, Ber. Dtsch. Chem. Ges., 1902, 35, 302 CrossRef CAS PubMed.
- CCDC 1415494, compound (6).
- J. Labuta, J. P. Hill, S. Ishihara, L. Hanyková and K. Ariga, Acc. Chem. Res., 2015, 48, 521 CrossRef CAS PubMed.
- A. Shundo, J. Labuta, J. P. Hill, S. Ishihara and K. Ariga, J. Am. Chem. Soc., 2009, 131, 9494 CrossRef CAS PubMed.
- J. Labuta, S. Ishihara, T. Šikorský, Z. Futera, A. Shundo, L. Hanyková, J. V. Burda, K. Ariga and J. P. Hill, Nat. Commun., 2013, 4, 2188 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental data and characterisation for all compounds. CCDC 1415494. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc06306j |
| This journal is © The Royal Society of Chemistry 2015 |