
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Steric C–N bond activation on the dimeric macrocycle [{P(μ-NR)}2(μ-NR)]2†
Yan X.
Shi
a,
Rong Z.
Liang
a,
Katherine A.
Martin
a,
Daniel G.
Star
b,
Jesús
Díaz
c,
Xin Y.
Li
a,
Rakesh
Ganguly
a and
Felipe
García
*a
aSchool of Physical and Mathematical Sciences, Division of Chemistry and Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore. E-mail: fgarcia@ntu.edu.sg
bChemistry Department, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
cDepartamento de Química Orgánica e Inorgánica, Facultad de Veterinaria, Universidad de Extremadura, Cáceres 10071, Spain
First published on 28th August 2015
Abstract
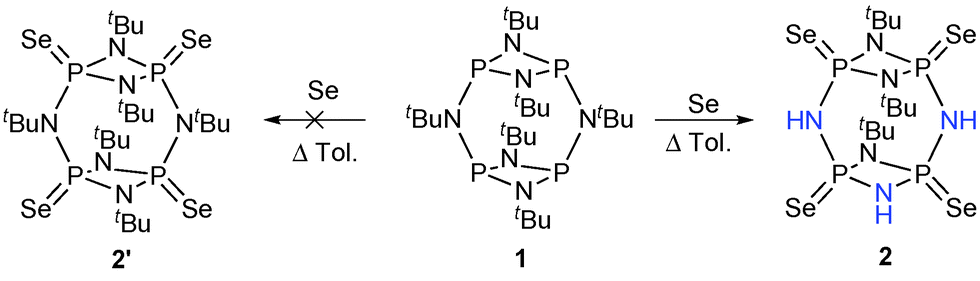
Dimeric cyclophosphazanes [{P(μ-NR)}2(μ-NR)]2 [R = tBu ( 1) and iPr ( 3)] were oxidized with elemental selenium. During these reactions an unexpected C–N bond cleavage and N–H bond formation occurred. Compound 1 produced P4(μ-NtBu)3(μ-NH)3Se4 ( 2) where three tBu groups were lost in the form of isobutylene. In contrast, during the oxidation of the less sterically hindered 3, the resulting product, P4(μ-NiPr)5(μ-NH)Se4 ( 4), showed only one substituent loss. Theoretical studies confirmed the steric nature of the driving force underlying the different outcomes.
Cyclophosph(III/III)azanes, with a variety of substituents, have been extensively studied over the past decades.1 One of their main features is that they can be found in a myriad of topological arrangements spanning from simple dimeric units to cage2 and macrocyclic arrangements.3–5 Moreover, the facile functionalization of dimeric rings of the type [ClP(μ-NR)]2 with a variety of nucleophiles6 lends these species unprecedented versatility as building blocks towards the synthesis of a wide range of multidentate ligands to transition metals.7,8 However, the intrinsic bond lability of these species renders them incapable of withstanding standard reaction conditions required for many simple organic transformations. Furthermore, an increased stability of the P–N framework upon oxidation of the phosphorus center has been reported.9 For example, Balakrishna et al. reported the synthesis of a series of cyclophosph(V/V)azanes showing enhanced air and moisture stability.10 These results prompted us to investigate the stability of cyclic cyclophosphazane cage frameworks of the type [{P(μ-NR)}2(μ-NR)]2 [R = tBu ( 1) and iPr ( 3)] upon oxidation. Herein, we report the unexpected C–N bond activation that occurs upon oxidation of the dimeric cyclophosphazanes [{P(μ-NtBu)}2(μ-NtBu)]2 ( 1) with elemental Se. Compound 1, was previously synthesized via the condensation reaction between [ClP(μ-NtBu)]2 and [LiNtBuP(μ-NtBu)]2, and was isolated as co-crystals of [LiNtBuP(μ-NtBu)]21 (1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) by Chivers et al.11 Our initial work focused on the purification of 1, which was successfully crystalized in THF at room temperature.
:1 ratio) by Chivers et al.11 Our initial work focused on the purification of 1, which was successfully crystalized in THF at room temperature.
Oxidation of 1 with elemental Se (molar ratio of 1:4.4) was performed in refluxing toluene for 72 hours. In situ31P{1H} NMR spectroscopic studies were used to monitor the reaction mixture. The expected fully oxidized and symmetrical product, [{P(Se)(μ-NtBu)}2(μ-NtBu)]2 ( 2′), would display a single resonance within the range of δ 0–50 ppm (cf., 117.2 ppm in 1) consistent with previously reported cyclophosph(V/V)azane selenide derivatives (e.g., [{P(Se)(μ-NtBu)}2{1,3-(O)2-C6H4}]2 and cis-[Cy(H)NP(Se)(μ-NCy)]2, δ 32.7 and δ 35.4 ppm, respectively).12,13 To our surprise, after 36 hours of reflux, the in situ31P{1H} NMR spectrum showed a neat incipient set of signals composed of two groups of multiple resonances at δ ∼24 and ∼22 ppm, respectively. In situ31P{1H} NMR spectrum taken after refluxing for a further 36 hours confirmed reaction completion. The reaction mixture was filtered through Celite, and the filtrate dried off under vacuum. Crystallization of the residue in toluene afforded crystals of compound 2 suitable for X-ray crystallographic studies (Scheme 1).
 | ||
| Scheme 1 Synthesis of compound 2. | ||
The X-ray solid state structure of P4(μ-NtBu)3(μ-NH)3Se4 ( 2) shows a tetraselenide-oxidized product where three tert-butyl groups, two on the ring-bridging nitrogen atoms and one on the P2N2 ring, were cleaved off as is shown in Fig. 1.
 | ||
| Fig. 1 ORTEP diagram for 2. Selected bond lengths [Å] and angles [deg]: N(1)–P(2) 1.717(5), N(1)–P(1) 1.682(5), N(2)–P(2) 1.723(5), N(2)–P(1) 1.700(5), N(3)–P(3) 1.716(4), N(3)–P(4) 1.675(5), N(5)–P(2) 1.604(5), N(5)–P(3) 1.613(5), Se(1)–P(1) 2.085(16), Se(2)–P(2) 2.102(16), N(1)–P(1)–Se(1) 121.8(18), N(2)–P(1)–Se(1) 118.6(17), N(6)–P(1)–Se(1) 109.3(17), N(1)–P(2)–Se(2) 115.1(17), N(2)–P(2)–Se(2) 116.3(18), N(5)–P(3)–Se(3) 109.3(18), N(6)–P(4)–Se(4) 107.5(17). Ellipsoids are drawn at 50% probability. H atoms have been omitted for clarity. | ||
The cleavage of N–tBu bonds resulted in the formation of three new N–H bonds. The 31P{1H} NMR spectrum of 2 shows two second order multiplets centred at δ 24.68 and 22.08 ppm, respectively, corresponding to the two distinct phosphorus environments present. The three remaining tBu groups in 2 can be observed in the 1H NMR spectrum as two singlet resonances with an intensity ratio of 1:2 at δ 1.31 and 1.62 ppm (cf., 1.57 and 1.53 ppm in 1). Furthermore, the 1H NMR spectrum displays signals resulting from the newly formed N–H bonds in the form of two broad singlets at δ 5.64 and 4.28 ppm, with an intensity ratio of 2:1, which correspond to the protons on the ring-bridging nitrogen and the P2N2 ring, respectively. This observation is also consistent with typical NH absorptions bands observed at 3192 cm−1 in the IR spectrum.
The mean P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se bond distance in 2, 2.0897(16) Å, is comparable with previously reported analogues (cf. 2.0913 in cis-[(μ-NtBu)2(P(Se)NC4H8NMe)2]). The P–N average bond distances within the P2N2 ring and the P–N bridging groups are 1.700(5) and 1.651(5) Å (cf.1, 1.791 and 1.763 Å, respectively).11 Upon oxidation, the P2N2 rings exhibit less acute angles around the nitrogen atoms as seen in analogous oxidized cyclophosph(III/III)azanes. These observations may be rationalized by the absence of sterically demanding lone pairs and greater inductive effect of the P(V) atoms, and the presence of fewer tert-butyl groups.
Se bond distance in 2, 2.0897(16) Å, is comparable with previously reported analogues (cf. 2.0913 in cis-[(μ-NtBu)2(P(Se)NC4H8NMe)2]). The P–N average bond distances within the P2N2 ring and the P–N bridging groups are 1.700(5) and 1.651(5) Å (cf.1, 1.791 and 1.763 Å, respectively).11 Upon oxidation, the P2N2 rings exhibit less acute angles around the nitrogen atoms as seen in analogous oxidized cyclophosph(III/III)azanes. These observations may be rationalized by the absence of sterically demanding lone pairs and greater inductive effect of the P(V) atoms, and the presence of fewer tert-butyl groups.
In order to gain insights into the reaction mechanism, the reaction was performed in a sealed Young's tap NMR tube. The in situ31P{1H} NMR spectra show the initial formation of a series of partially oxidized intermediates before the formation of compound 2. During the course of the reaction, the 1H NMR spectra show the appearance of two new signals with intensity ratio 1:3 at δ 4.76 and 1.64 ppm respectively, as the only detectable by-product. These two resonances were found to be in accordance with 1H NMR spectrum of isobutylene (Fig. 2, compound a).14
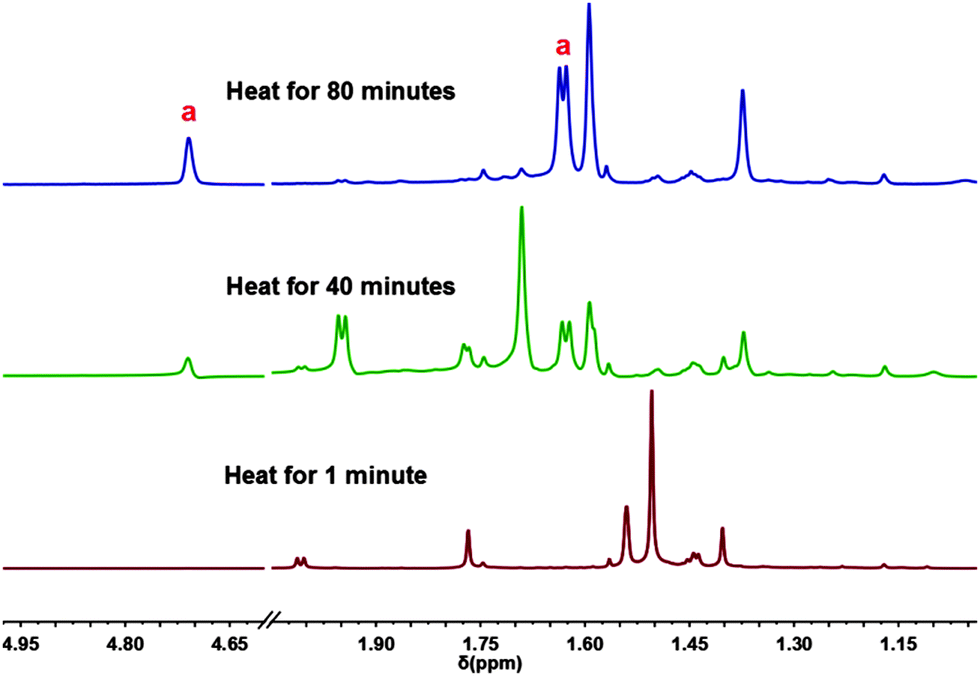 | ||
| Fig. 2 1H NMR spectrum of the reaction of P4(μ-NtBu)6 ( 1) with elemental Se in a sealed NMR tube under reflux in toluene-d8. | ||
Several unsuccessful attempts were made to isolate the partially oxidized transient species (i.e., 1a, 1b and 1c in Scheme 2, see ESI†); however a singlet at ∼25 ppm observed during the course of the reaction can be attributed to the fully-oxidized non-cleaved compound 2′. We observed that only once 2′ is detected in the 31P{1H}NMR spectra the signal set corresponding to isobutylene commence to appear in the 1H NMR spectra (see ESI†). To further evaluate the influence of stoichiometry and reaction conditions upon the obtainment of 2 and/or observed intermediates in isolatable form, 1 was oxidized in a 1:2.2 ratio under identical experimental conditions. Further to this, the use of alternative solvents such as THF and ACN was explored under reflux and at room temperature in both 1:4.4 and 1:2.2 molar ratios. In all cases a mixture of compound 2 and the di-oxidized derivative ( 1b) were observed in the in situ31P{1H} NMR spectrum after prolonged reaction times, suggesting that the cleaved product 2 is the most thermodynamically stable fully-oxidized product.
 | ||
| Scheme 2 | ||
Since the P4N6 framework contracts upon oxidation (vide supra), we presume that steric strain release is the driving force for the observed C–N bond cleavage. In order to validate this observation, the less sterically hindered dimeric phosphazane [{P(μ-NiPr)}2(μ-NiPr)]2 ( 3) was synthesized. In this context, is important to note that frameworks of the type [{P(μ-NR)}2(μ-NR)]2 are thermodynamically unstable with respect of their adamantoid isomeric forms [P4(NR)6]. In the case of compound 1, isomerization to an adamantoid structure is prevented by its highly sterically encumbered nature.15 Since compound 3 is less thermodynamically stable towards isomerization than 2, milder reaction conditions were explored, hence, compound 3 was refluxed in ACN for 24 h. The in situ31P{1H} NMR spectrum showed a mixture of products corresponding to partially oxidized species (i.e., 3a, 3b and 3c), minor products, and a signal pattern for P4(μ-NiPr)5(μ-NH)Se4 ( 4) consistent with one iPr group having being cleaved. The 31P{1H} and 1H NMR spectra show complex signals at 55.25 and 30.65 ppm and at 6.06 and 5.34 ppm (ratio 1:2, respectively) consistent with the proposed structure. The 1:2 ratio found for the NH groups in the 1H NMR spectrum (cf.2, 2:1 ratio) equal to the relative ratio of ring-bridging and internal P2N2 ring nitrogen atoms present in 3, indicates that a mixture of both terminal and P2N2 mono-cleaved structures are present. This was further corroborated by single crystal X-ray diffraction studies that show that one iPr group is cleaved off and is disordered over the six nitrogen atoms present within the P4N6 backbone of 4 (Fig. 3).
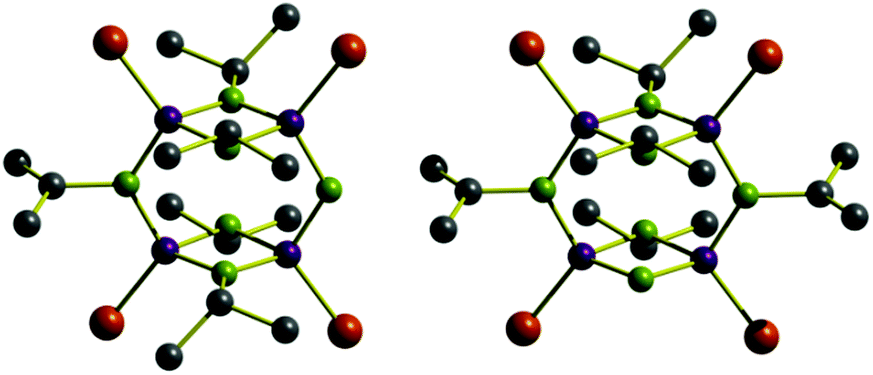 | ||
| Fig. 3 Structural representation of the two species present in 4. See ESI† for crystallographic data and ORTEP diagram. | ||
Similarly to 2, the mean P–N and PSe bond distances in 4 are 1.700(10) and 2.075(4) Å, respectively. The symmetric P2N2 rings are almost planar (puckered by 4.7°) and are virtually perpendicular to the macrocyclic plane (ca. 89°). Unfortunately, compound 4 readily decomposes upon isolation prohibiting any further characterization. Furthermore, despite several attempts, it was not possible to achieve the synthesis of analogues to 1 and 3 containing less bulky substituents (e.g., nPr, Et or Me). However, to rationalize the energetics of the reaction and further elucidate the observed C–N bond cleavage, DFT calculations were performed. An assessment of the relative thermodynamic stabilities the compounds can be obtained by directly comparing the enthalpies of reactants and products that share the same degree of oxidation (i.e., 2′ and 4′vs.2 and 4, respectively). Our studies indicate that in the case of tBu substituents, the loss of three substituents is largely favoured (−82.5 kcal mol−1) followed by the instance in which two groups are cleaved (−81.4 kcal mol−1). In contrast, for the less bulky iPr substituent only the C–N bond is energetically favoured (−0.8 kcal mol−1). To further illustrate the steric nature of the observed C–N bond activation we performed parallel studies for the hypothetical nPr substituted analogue. In this case, the loss of any number of substituents would result in an overall energy cost making it the most thermodynamically unfavourable of modelled systems. Considering solely thermodynamic control over the reaction products we can establish that the C–N bond cleavage is due to steric factors and is generated to ease tensions within the ring upon oxidation of the phosphorus centres. The DFT calculated trends are in agreement with the experimental data obtained for compounds 2 and 4 which underwent C–N cleavage of three and one substituents respectively (see ESI†).
Furthermore, geometry optimizations of the fully oxidized uncleaved product 2′ and 4′, reveal noticeable differences between the C–N bond distances for internal P2N2 ring and bridging nitrogen atoms (1.53 and 1.60 Å for 2′; 1.52 and 1.50 Å for 2′; respectively). Whilst bond distances within 1 and 3 follow the same trend, there is a greater discrepancy between P2N2 ring and bridging C–N bond lengths for the oxidized products than their unoxidized counterparts (0.07 Å in 2versus 0.04 Å in 1). Calculation of the energy differences associated with the cleavage of the nitrogen substituent at each of these two distinct positions showed that, as expected, the loss of substituents in bridging positions is more favourable than those bonded directly to the P2N2 rings (see ESI†). Our theoretical studies only account for relative stabilities of compounds 2 and 4 (and their subsequent cleavages) when compared with their corresponding uncleaved species 2′ and 4′. We acknowledge that this approach does not account for other important factors involved in this process such as entropy or thermal conditions of the reaction, Gibbs energies, etc. Such studies are currently underway. However, for the purpose of this work, our approach provided results that were consistent with our experimental observations with a feasible computational cost.
In summary, we have shown that the oxidation reaction of [{P(μ-NR)}2(μ-NR)]2, R = 1 (tBu) and 3 (iPr), with elemental Se prompts the activation of C–N bonds due to ring contraction resulting in their cleavage under mild experimental conditions. Our experimental and theoretical studies demonstrate that steric factors, most probably steric strain release, play a crucial role in the activation of the C–N bonds. The work reported here highlights the potential of steric bond activation and its implications with a broader scope for application to main group frameworks in general. The rational design of synthetic approaches, which fully exploit this approach in a wider range of main group frameworks, is an exciting challenge in the area of metal-free bond activation.
We would like to thank NTU (grant number: M4080552) for financial support. J. D. thanks COMPUTAEX for granting access to LUSITANIA supercomputing facilities.
Notes and references
- (a) T. Roth, H. Wadepohl, D. S. Wright and L. H. Gade, Chem. – Eur. J., 2013, 19, 13823–13837 CrossRef CAS PubMed; (b) K. C. K. Swamy, G. Gangadhararao, V. Srinivas, K. N. N. Bhuvan, E. Balaraman and M. Chakravarty, Inorg. Chim. Acta, 2011, 372, 374–382 CrossRef PubMed; (c) M. S. Balakrishna, D. Suresh and J. T. Mague, Inorg. Chim. Acta, 2011, 372, 259–265 CrossRef CAS PubMed; (d) M. S. Balakrishna, J. Organomet. Chem., 2010, 695, 925–936 CrossRef CAS PubMed.
- (a) A. Bashall, E. L. Doyle, F. Garcia, G. T. Lawson, D. J. Linton, D. Moncrieff, M. McPartlin and A. D. Woods, Chem. – Eur. J., 2002, 8, 5723–5731 CrossRef CAS; (b) F. Garcia, R. A. Kowenicki, L. Riera and D. S. Wright, Dalton Trans., 2005, 2495–2496 RSC.
- P. Kommana and K. C. K. Swamy, Inorg. Chem., 2000, 39, 4384–4385 CrossRef CAS.
- (a) K. W. Muir and J. F. Nixon, J. Chem. Soc., Chem. Commun., 1971, 1405–1406 RSC; (b) H. J. Chen, R. C. Haltiwanger, T. G. Hill, M. L. Thompson, D. E. Coons and A. D. Norman, Inorg. Chem., 1985, 24, 4725–4730 CrossRef CAS; (c) P. B. Hitchcock, M. F. Lappert and M. Layh, J. Organomet. Chem., 1997, 529, 243–255 CrossRef CAS.
- (a) F. García, R. A. Kowenicki, I. Kuzu, L. Riera, M. McPartlin and D. S. Wright, Dalton Trans., 2004, 2904–2909 RSC; (b) F. Dodds, F. García, R. A. Kowenicki, M. McPartlin, A. Steiner and D. S. Wright, Chem. Commun., 2005, 3733–3735 RSC; (c) F. Dodds, F. Garcia, R. A. Kowenicki, M. McPartlin, L. Riera, A. Steiner and D. S. Wright, Chem. Commun., 2005, 5041–5043 RSC; (d) F. Dodds, F. Garcia, R. A. Kowenicki, S. P. Parsons, M. McPartlin and D. S. Wright, Dalton Trans., 2006, 4235–4243 RSC; (e) A. Bashall, E. L. Doyle, C. Tube, S. J. Kidd, M. McPartlin, A. D. Woods and D. S. Wright, Chem. Commun., 2001, 2542–2543 RSC; (f) F. García, R. A. Kowenicki, I. Kuzu, M. McPartlin, L. Riera and D. S. Wright, Inorg. Chem. Commun., 2005, 1060–1062 CrossRef PubMed.
- (a) R. Keat, D. S. Rycroft and D. G. Thompson, J. Chem. Soc., Dalton Trans., 1979, 1224–1230 RSC; (b) G. S. Ananthnag, S. Kuntavalli, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2012, 51, 5919–5930 CrossRef CAS PubMed; (c) A. D. Woods and M. McPartlin, Dalton Trans., 2004, 90–93 RSC; (d) M. Rastätter, P. W. Roesky, D. Gudat, G. B. Deacon and P. C. Junk, Chem. – Eur. J., 2007, 13, 7410–7415 CrossRef PubMed; (e) R. Kuzora, A. Schulz, A. Villinger and R. Wustrack, Dalton Trans., 2009, 9304–9311 RSC; (f) Z. Zak and T. Glowiak, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 445–446 CrossRef.
- (a) A. Bashall, A. D. Bond, E. L. Doyle, F. García, S. Kidd, G. T. Lawson, M. C. Parry, M. McPartlin, A. D. Woods and D. S. Wright, Chem. – Eur. J., 2002, 8, 3377–3385 CrossRef CAS; (b) O. J. Scherer, R. Anselmann, R. T. Paine and S. Karthikeyan, Inorganic Syntheses, John Wiley & Sons, Inc., 2007, pp. 7–12 Search PubMed; (c) M. Vijjulatha, K. C. K. Swamy, J. J. Vittal and L. L. Koh, Polyhedron, 1999, 18, 2249–2254 CrossRef CAS; (d) K. V. Axenov, V. V. Kotov, M. Klinga, M. Leskelä and T. Repo, Eur. J. Inorg. Chem., 2004, 695–706 CrossRef CAS PubMed.
- M. S. Balakrishna, R. Venkateswaran and J. T. Mague, Inorg. Chem., 2009, 48, 1398–1406 CrossRef CAS PubMed.
- Y. X. Shi, R. Z. Liang, K. A. Martin, N. Weston, S. Gonzalez-Calera, R. Ganguly, Y. Li, Y. Lu, A. J. M. Ribeiro, M. J. Ramos, P. A. Fernandes and F. García, Inorg. Chem., 2015, 54, 6423–6432 CrossRef CAS PubMed.
- (a) A. Nordheider, T. Chivers, R. Thirumoorthi, I. Vargas-Baca and J. D. Woollins, Chem. Commun., 2012, 48, 6346–6348 RSC; (b) P. Chandrasekaran, J. T. Mague and M. S. Balakrishn, Eur. J. Inorg. Chem., 2011, 2264–2272 CrossRef CAS PubMed; (c) M. S. Balakrishna, D. Suresh and J. T. Mague, Eur. J. Inorg. Chem., 2010, 4201–4210 CrossRef CAS PubMed; (d) T. Chivers, M. Krahn, M. Parvez and G. Schatte, Inorg. Chem., 2001, 40, 2547–2553 CrossRef CAS PubMed; (e) P. Kilian, P. Pazdera, J. Marek, J. Novosad and J. Touzin, Z. Anorg. Allg. Chem., 1998, 624, 1497–1502 CrossRef CAS.
- J. K. Brask, T. Chivers, M. L. Krahn and M. Parvez, Inorg. Chem., 1999, 38, 290–295 CrossRef CAS.
- G. S. Ananthnag, S. Kuntavalli, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2012, 51, 5919–5930 CrossRef CAS PubMed.
- P. Chandrasekaran, J. T. Mague and M. S. Balakrishna, Eur. J. Inorg. Chem., 2011, 2264–2272 CrossRef CAS PubMed.
- A. J. Kell, A. Alizadeh, L. Yang and M. S. Workentin, Langmuir, 2005, 21, 9741–9746 CrossRef CAS PubMed.
- (a) O. J. Scherer, K. Andres, C. Krüger, Y.-H. Tsay and G. Wolmerhäuser, Angew. Chem., 1980, 92, 563–564 ( Angew. Chem., Int. Ed. Engl. , 1980 , 19 , 571–572 ) CrossRef CAS PubMed; (b) T. G. Hill, R. C. Haltiwanger, M. L. Thompson, S. A. Katz and A. D. Norman, Inorg. Chem., 1994, 33, 1770–1777 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and spectral data, and crystallographic data. CCDC 1407770–1407773. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc06034f |
| This journal is © The Royal Society of Chemistry 2015 |