
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstituted ferrocenes and iodine as synergistic thermoelectrochemical heat harvesting redox couples in ionic liquids†
E. H. B.
Anari
a,
M.
Romano
b,
W. X.
Teh
a,
J. J.
Black
a,
E.
Jiang
a,
J.
Chen
b,
T. Q.
To
a,
J.
Panchompoo
a and
L.
Aldous
*a
aSchool of Chemistry, UNSW Australia, Sydney, NSW 2052, Australia. E-mail: l.aldous@unsw.edu.au
bIntelligent Polymer Research Institute, Innovation Campus, University of Wollongong, Wollongong, NSW 2522, Australia
First published on 4th November 2015
Abstract
Combining ferrocene and iodine results in enhanced thermoelectrochemical (or thermogalvanic) waste heat harvesting abilities, for both the Seebeck coefficient and the overall power output. All systems displayed a mixture of ferrocene, ferrocenium, iodine and triiodide. The observed enhancement correlates with lower electron-density on the ferrocene; the synergistic improvement observed for mixtures of substituted ferrocenes and iodine is attributed to the formation of charge-transfer complexes. Combining dibutanoylferrocene and iodine resulted in the highest Seebeck coefficient of 1.67 mV K−1.
Worldwide, heat is converted into electricity on a tremendous scale. The Seebeck effect allows arguably the simplest process, whereby a difference in temperature can be converted directly into electrical power with no moving parts.1 This simplicity has made it a hallmark energy system of unmanned space exploration.1 The majority of thermoelectric systems are based upon solid-state semiconductor devices, typically containing rare metalloids such as Bi2Te3,2 and which suffer from rigidity, expense and low efficiencies.1
Thermoelectrochemical cells containing a liquid electrolyte with a suitable redox couple can also generate electrical power from a temperature difference. These systems offer many advantages such as flexibility,3 low cost4 and are suited to the harvesting of low-grade heat.5
The Seebeck coefficient (Se, eqn (1)) expresses the potential difference (ΔE) generated across two electrodes sharing a common electrolyte, as a function of their temperature difference (ΔT).6 Dissolved redox couples at different temperatures will have a free-energy difference proportional to the entropy difference between the redox states (ΔS). Unlike in semi-conductors, these liquid, solid or gaseous electrochemical systems give rise to significant Se values on the order of mV K−1.7 This is important, as the maximum power is directly proportional to the Se value, and the efficiency of heat conversion is proportional to the Se squared.8
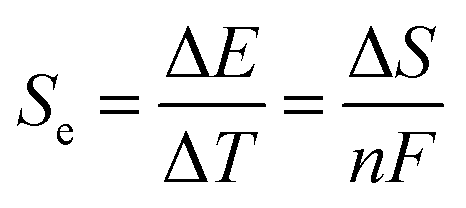 | (1) |
The majority of studies into thermoelectrochemical cells have employed aqueous electrolytes containing inorganic redox couples. A prime example is aqueous potassium ferrocyanide/ferricyanide solutions, which exhibit a relatively large Se on the order of ca. −1.4 mV K−1.3,4 Short-lived systems can exceed this, recently reaching +13.6 mV K−1.9 However, the volatility of water can be a major limitation for thermoelectrochemical cells and so ionic liquids (ILs) have been proposed as non-volatile electrolytes for thermoelectrochemical cells.5 IL-based thermoelectrochemical cells can be operated at temperatures above 100 °C with no degradation, as a result of the high thermal stability and the low (near-zero) vapour pressure of many ILs.5a ILs are known to be highly structured liquids, and entropy is highly significant in many processes in ILs.10 In 2013 a Co(II)/Co(III) complex reached Se values of 1.40 to 1.88 mV K−1 in a range of ILs.5b
Despite progress, relying upon the entropy change inherent in a single redox process is clearly self-limiting; too great an entropy change generally correlates with lower exchange current densities and necessitates an electrocatalytic material. Electrocatalysis in ILs is a relatively new and poorly understood area.11 In an effort to move beyond this limiting factor, this work investigates combining two redox couples based upon ferrocene and iodine. When employed individually they represent poor thermoelectrochemical redox couples, but together they display a synergistically enhanced performance. This opens new routes to maximising performance by overcoming the limitations noted above.
Both the ferrocene|ferrocenium12 and iodide|triiodide13 redox couples find extensive uses in electrochemistry, and have been (separately) well characterised in ILs. Both ferrocenium-based ILs14 and iodide/triiodide-based ILs13 are known. Recently, combining redox couples has been beneficially employed in IL-based dye sensitised solar cells.15
In this work, redox couples were investigated by dissolving individual redox couples (ferrocene|ferrocenium and iodide|triiodide) and also by mixing ferrocene and iodide, which are in equilibrium with their ferrocenium triiodide salt (eqn (2)). The reaction between ferrocene and iodide has been the subject of extensive fundamental research relating to equilibrium constants and kinetics,16 although not in ILs.
Fc + ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) I2 ⇌ Fc+ + [I3]− I2 ⇌ Fc+ + [I3]− | (2) |
In addition to the ferrocenium triiodides being inherently electroactive, the overall equilibrium is also temperature-sensitive. While ferrocenium species rapidly decomposes in oxygenated organic solvents,17 they were found to be stable when dissolved in the ionic liquid 1-ethyl-3-methyl-imidazolium bis(trifluoromethylsulfonyl)imide ([Emim][NTf2]), hence all studies were performed in this IL.
Several substituted ferrocenes were synthesised (full details in ESI†). They are ferrocene (Fc), acetylferrocene (AcFc), ethylferrocene (EtFc), dibutylferrocene (DiBuFc) and dibutanoylferrocene (DiBoylFc).
The Seebeck coefficients (Se) were measured in a U-shaped assembly (U-cell, full details in the ESI†) using platinum electrodes. Fig. 1(a) displays the potential difference of 15 mM Fc/15 mM Fc+ (▲), 15 mM I−/15 mM [I3]− (●) and 30 mM “[Fc][I3]” (from 30 mM Fc and 45 mM I2, ■) in [Emim][NTf2], measured as a function of temperature; the corresponding Se values are summarised in Table 1.
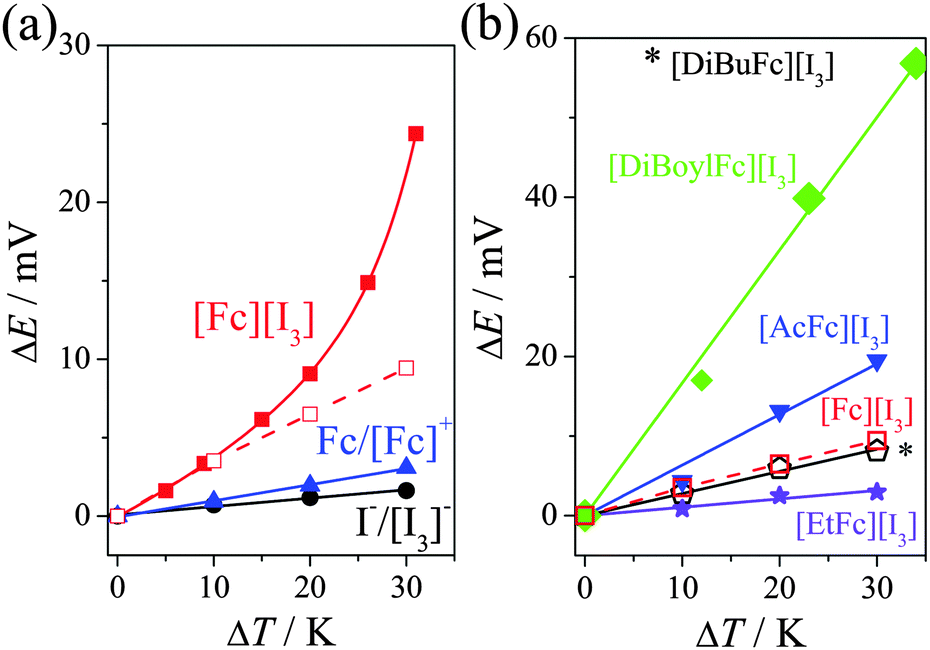 | ||
Fig. 1 (a) Potential difference recorded as a function of temperature for 15 mM Fc/15 mM Fc+ (●), 15 mM I−/15 mM [I3]− ( ) and 30 mM of [Fc][I3] ( ) and 30 mM of [Fc][I3] ( in U-tube, in U-tube,  in battery casing) in [Emim][NTf2]. Also (b) potential difference for 30 mM of [Fc][I3] ( in battery casing) in [Emim][NTf2]. Also (b) potential difference for 30 mM of [Fc][I3] ( ) 30 mM [AcFc][I3] ( ) 30 mM [AcFc][I3] ( ), 30 mM [EtFc][I3] ( ), 30 mM [EtFc][I3] ( ), 30 mM [DiBuFc][I3] (★) and 30 mM [DiBoylFc][I3] ( ), 30 mM [DiBuFc][I3] (★) and 30 mM [DiBoylFc][I3] ( ) in [Emim][NTf2]. Tcold = 20 °C. Error bars are smaller than data points. ) in [Emim][NTf2]. Tcold = 20 °C. Error bars are smaller than data points. | ||
| Redox couple in [Emim][NTf2] | Seebeck coefficient/mV K−1 (value from CR2032 casing) |
|---|---|
| 15 mM Fc/15 mM [Fc]+ | 0.100 ± 0.002 |
| 15 mM I−/15 mM [I3]− | 0.057 ± 0.003 |
| 30 mM [Fc][I3] | 0.813* (0.291 ± 0.002) |
| 30 mM [EtFc][I3] | 0.106 ± 0.008 |
| 30 mM [DiBuFc][I3] | 0.279 ± 0.006 |
| 30 mM [AcFc][I3] | 0.636 ± 0.033 |
| 30 mM [DiBoylFc][I3] | 1.669 ± 0.045 |
The value measured here for Fc|Fc+ is essentially identical to the value reported by Migita et al. (+0.10 ± 0.01 mV K−1).18 The measured value for I−|[I3]− is slightly lower than that reported by Abraham et al. (+0.154 mV K−1 in [Emim][NTf2]5a) although those samples were ca. 30 fold more concentrated.
The [Fc][I3] system is more complex, and interestingly displayed a non-linear potential vs temperature trend. The [Fc][I3] also displayed consistently higher potential values that that of the individual ferrocene and iodine systems. The non-linear trend was present during heating and cooling, displaying no hysteresis, although it is important to note that it took ca. 1 h for these values to stabilise.
A maximum potential of ca. 24.4 mV was achieved by [Fc][I3] at ΔT = 30 K, which equates to an Se of ca. 0.83 mV K−1. Therefore the simple expedient of mixing Fc0 with I2 results in an Se value which (at ΔT = 30 K) equates to a 14-fold increase over I−/[I3]− alone, and a 5-fold increase over the sum of the individual Fc/Fc+ and I−/[I3]− redox couples, representing a significant synergy when the two are mixed to form [Fc][I3]. Further increases in temperature resulted in some instability due to gradual evaporation of the volatile materials from the open U-tube, and at ca. 100 °C [Fc][I3] underwent irreversible decomposition to form iron iodide.
The same systems were investigated in a platinum-coated CR2032 battery casing, which provides 2 mm electrode separation (cf. 2 cm in the U-tube) and is more suited for power generation. The hermetically sealed casing also allowed higher temperature differences without any loss of the volatile redox compounds. In this case, the [Fc][I3] initial trend matched that of the U-tube but deviated at higher temperatures (Se = 0.291 ± 0.002 mV K−1, hollow symbols in Fig. 1(a)). Nevertheless, the Se value still displayed the synergy discussed above. The Se value of Fc/Fc+ was also measured in the battery casing and found to be 0.081 ± 0.001 mV K−1 (cf. 0.100 ± 0.002 mV K−1 in the U-tube), and all systems gave ca. 20% lower Seebeck coefficients in the battery casing. This is predominately due to a temperature drop from the heated exterior to the interior of the cell. In the U-tube (electrodes separated by 2 cm) it took ca. 1 h for the potential values to stabilise, whereas in the battery casing (2 mm separation) it took only a few minutes (Fig. S3, ESI†). In the battery casing there is more significant thermal convection, whereas in the U-tube the temperature-induced change in the equilibrium in eqn (2) appears to become more significant over time. This mirrors observations in aqueous systems where varying the electrode separation and electrode orientation to gravity can either increase or decrease power according to the precise redox couple utilised.19
The power discharge characteristics of the systems were also investigated in the battery casing. As demonstrated in Fig. 2, 30 mM [Fc][I3] generated significantly more power than 30 mM Fc|Fc+, largely due to the higher Se value of the former. The precise charge carrier(s) involved in the [Fc][I3] system is currently unknown. The power also refers to the steady-state conditions obtained after 10 min discharge. Given their differing diffusion coefficients12,13 and the possibility for both species to undergo Grotthuss-like conductivity, further work is required to investigate what happens to these systems under extended discharge conditions.
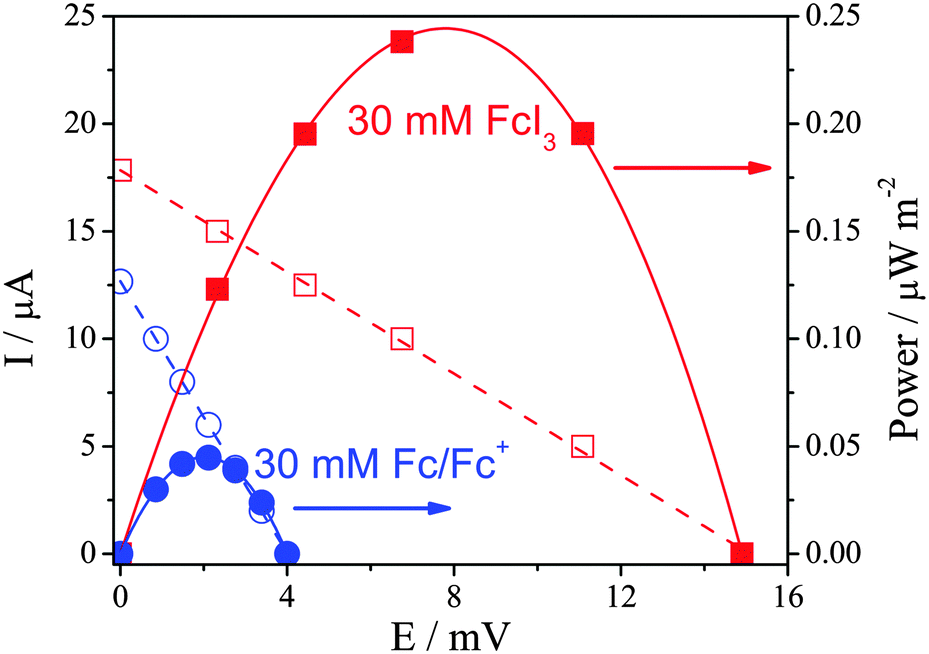 | ||
Fig. 2 Current–potential discharge trends, displaying power generated by 30 mM Fc/Fc+ (15 mM of each) ( ) and 30 mM of [Fc][I3] ( ) and 30 mM of [Fc][I3] ( ) in [Emim][NTf2] in a Pt-coated CR2032 battery casing at ΔT = 50 °C (Tcold = 30 °C). ) in [Emim][NTf2] in a Pt-coated CR2032 battery casing at ΔT = 50 °C (Tcold = 30 °C). | ||
While the temperature-dependant equilibrium in the ferrocenium triiodide salt clearly has some influence, it does not explain the observed synergy resulting in the Se increases. To investigate further, the ferrocene core was systematically altered by adding electron-donating (alkyl) or electron-withdrawing (alkanoyl) groups. Fig. 1(b) displays the potential differences measured for 45 mM I2 and 30 mM of Fc (□), AcFc (▼), EtFc (●), DiBuFc (★) and DiBoylFc (◆) in [Emim][NTf2]. Table 1 lists the Se values of all redox couple species studied in this work, and it was found that 30 mM “[DiBoylFc][I3]” possesses the highest Se value of 1.67 ± 0.05 mV K−1.
The trend in Se values follows the order DiBoylFc > AcFc > Fc > DiBuFc > EtFc, which follows the anticipated electron density on the ferrocene centre from lowest to highest. The inversion of DiBuFc and EtFc is an exception to this trend, and is likely due to a subtle contribution from the alkyl chains to the overall entropy change.
In organic solvents, electron-donating groups on the ferrocene moiety (e.g. alkyl groups) shifts the equilibrium in eqn (2) towards the right, resulting in more triiodide product formed.16a,b This was investigated in [Emim][NTf2] by cyclic voltammetry (CV), and the oxidation potentials of the substituted ferrocenes followed the expected trend in the IL (Fig. S5, ESI†). Comparison of the potentials relative to those of I−/[I3]− (Fig. S6, ESI†) and use of the Nernst equation allows us to estimate (see ESI† for full discussion) that a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture of DiBuFc and I2 should exist as 99.9% [DiBuFc][I3] (at 298 K); for DiBoylFc only ca. 10% exists as [DiBoylFc][I3]. However, when ferrocene and iodine were investigated in the same system the situation was more complex. Fig. 3 displays CVs of 15 mM DiBoylFc, 22.5 mM I2, and 15 mM [DiBoylFc][I3]; similar data for [Fc][I3] and [DiBuFc][I3] are included in the ESI.† When mixed, the I−/[I3]− feature displayed a ca. 50 mV anodic shift, the [I3]/I2 process was unchanged, while the DiBoylFc/[DiBoylFc]+ peaks shifted ca. 50 mV cathodically. Additionally, currents for the mixed ferrocenium triiodide systems were higher than expected, demonstrating some ‘nonadditivity’ of faradaic currents; this has been previously noted when ferrocene is mixed with cobaltocenium in ionic liquids.20
:3 mixture of DiBuFc and I2 should exist as 99.9% [DiBuFc][I3] (at 298 K); for DiBoylFc only ca. 10% exists as [DiBoylFc][I3]. However, when ferrocene and iodine were investigated in the same system the situation was more complex. Fig. 3 displays CVs of 15 mM DiBoylFc, 22.5 mM I2, and 15 mM [DiBoylFc][I3]; similar data for [Fc][I3] and [DiBuFc][I3] are included in the ESI.† When mixed, the I−/[I3]− feature displayed a ca. 50 mV anodic shift, the [I3]/I2 process was unchanged, while the DiBoylFc/[DiBoylFc]+ peaks shifted ca. 50 mV cathodically. Additionally, currents for the mixed ferrocenium triiodide systems were higher than expected, demonstrating some ‘nonadditivity’ of faradaic currents; this has been previously noted when ferrocene is mixed with cobaltocenium in ionic liquids.20
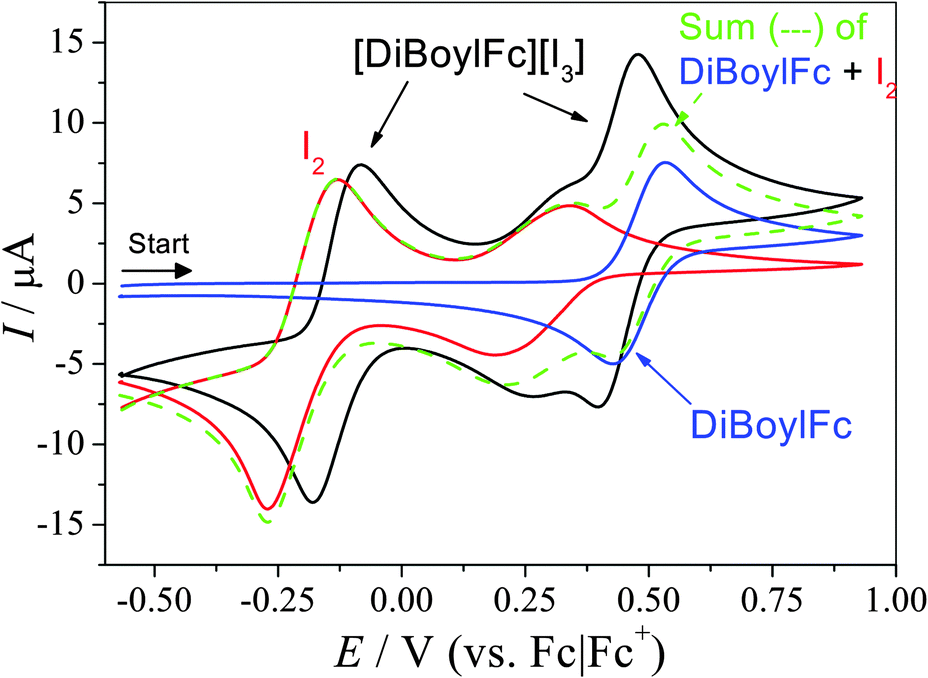 | ||
| Fig. 3 CVs recorded for 15 mM DiBoylFc, 22.5 mM I2, the additive sum of these two scans, and 15 mM [DiBoylFc][I3]. Scans recorded at a 1.6 mm Pt electrode, 293 K, 50 mV s−1, and corrected to the Fc|Fc+ formal potential. | ||
Further demonstration of this non-linear behaviour was obtained by UV-Vis spectroscopy. Ferrocene-iodine charge transfer complexes are known in non-aqueous solvents,16a and extinction coefficient of I2 and [I3]− are extremely structure- and solvation-sensitive.21Fig. 4(a) displays the titration of I2 with I− (introduced as [Emim]I), yielding the characteristic absorption bands.16d,22 Stoichiometric quantities of I2 and I− resulted in quantitative formation of [I3]−, as represented by the absorption maxima. Fig. 4(b) displays the same spectra of I2 and I2/I−, overlaid with the spectra for Fc/[Fc][PF6] and all five “ferrocenium triiodide” salts (as 3 mM ferrocene and 4.5 mM I2). All five ferrocene/iodine mixtures should have an absorbance below that of 3 mM [I3]−, as they will introduce 3 mM [I3]− or less, based upon the equilibrium in eqn (2). As the absorbance of the ferrocenium triiodides exceed this in every case, the solvation environment of [I3]− in the IL is clearly altered upon introduction of ferrocene species.
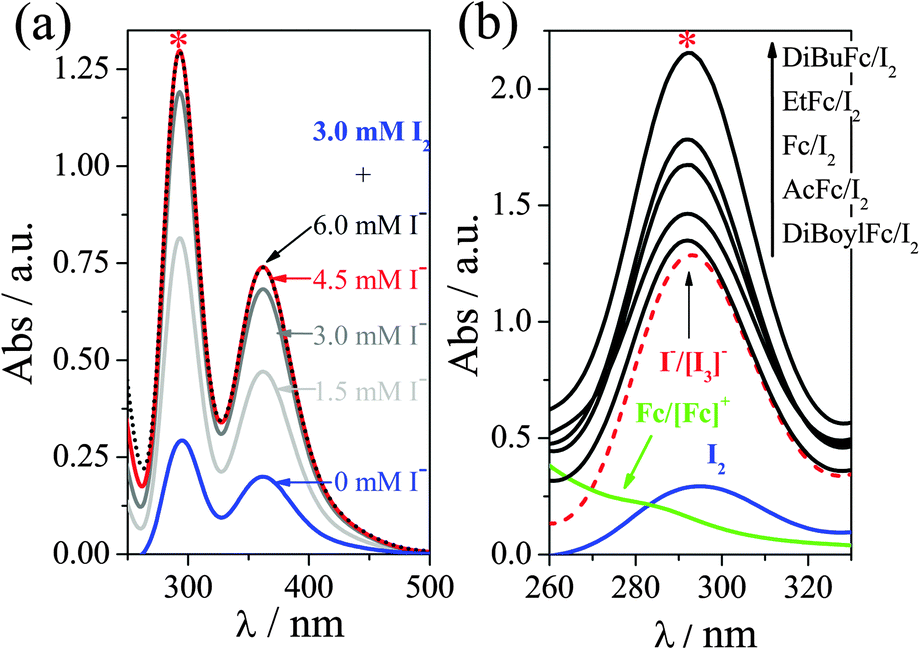 | ||
Fig. 4 UV-Vis absorption spectra of (a) 3 mM I2 in presence of various [Emim]I concentrations, and (b) 3 mM I2, 3 mM Fc/3 mM Fc+, 3 mM I−/3 mM [I3]− ( ) as well as 3 mM of the 5 [ferrocenium][I3] samples. All in [Emim][NTf2]. Pathlength 1 mm. ) as well as 3 mM of the 5 [ferrocenium][I3] samples. All in [Emim][NTf2]. Pathlength 1 mm. | ||
An interaction between ferrocene and iodine species will result in an additional entropic change upon a redox process occurring, relative to the individual species. These significant interactions can therefore explain the synergistic enhancement apparent in the Se coefficient. It is logical that the electron-deficient ferrocenes would display more significant charge-transfer complexation with the electron-rich and highly polarisable iodine species. This increased interaction mirrors the observed synergistic enhancement in the Se coefficient, with DiBoylFc predicted to interact with iodine the most and also displaying the highest Se coefficient of 1.67 mV K−1. This Se coefficient compares favourably with the highest Se value ever reported in the IL [Emim][NTf2], which was ca. 1.66 ± 0.02 mV K−1 for 100 mM [CoII/III[bpy]3][NTf2]2/3.5b The reported [CoII/III[bpy]3][NTf2]2/3 was deliberately chosen to result in the most significant entropy change upon changing oxidation state, with purely the cationic component contributing. In the case of the ferrocenium triiodides, we have demonstrated that two common, widely available, relatively poor thermoelectrochemical redox mediators (by conventional knowledge and approaches) can be combined to result in an excellent system when combined.
In conclusion, this work has investigated the thermoelectrochemistry of mixed redox couples (ferrocene and iodine) for the first time. These two couples were found to interact even in dilute solution, resulting in a synergistic thermoelectrochemical response. This likely stems from observed charge-transfer complex formation, which must introduce additional entropic changes when one component undergoes a redox process, additional to simple restructuring of the surrounding ionic liquid. This work therefore represents a new approach towards the development of thermoelectrochemical systems, with the value of 1.67 mV K−1 for [DiBoylFc][I3] being one of the highest reported Se value for a solute in the ionic liquid [Emim][NTf2].
The Australian Research Council (ARC DECRA DE130100770) is acknowledged for funding. The authors would like to thank the Australian National Fabrication Facility (ANFF) for equipment access.
Notes and references
- C. B. Vining, Nat. Mater., 2009, 8, 83–85 CrossRef CAS PubMed.
- D. M. Rowe, Thermoelectrics handbook: macro to nano, CRC/Taylor & Francis, Boca Raton, 2006 Search PubMed.
- M. S. Romano, N. Li, D. Antiohos, J. M. Razal, A. Nattestad, S. Beirne, S. Fang, Y. Chen, R. Jalili, G. G. Wallace, R. Baughman and J. Chen, Adv. Mater., 2013, 25, 6602–6606 CrossRef CAS PubMed.
- R. Hu, B. A. Cola, N. Haram, J. N. Barisci, S. Lee, S. Stoughton, G. Wallace, C. Too, M. Thomas, A. Gestos, M. E. Cruz, J. P. Ferraris, A. A. Zakhidov and R. H. Baughman, Nano Lett., 2010, 10, 838–846 CrossRef CAS PubMed.
- For example: (a) T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Chem. Commun., 2011, 47, 6260–6262 RSC; (b) T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Energy Environ. Sci., 2013, 6, 2639–2645 RSC; (c) V. Zinovyeva, S. Nakamae, M. Bonetti and M. Roger, ChemElectroChem, 2014, 1, 426–430 CrossRef; (d) M. Bonetti, S. Nakamae, B. T. Huang, T. J. Salez, C. Wiertel-Gasquet and M. Roger, J. Chem. Phys., 2015, 142, 244708 CrossRef PubMed.
- T. I. Quickenden and Y. Mua, J. Electrochem. Soc., 1995, 142, 3985–3994 CrossRef CAS.
- A. Gunawan, C. H. Lin, D. A. Buttry, V. Mujica, R. A. Taylor, R. S. Prasher and P. E. Phelan, Nanoscale Microscale Thermophys. Eng., 2013, 17, 304–323 CrossRef CAS.
- B. Burrows, J. Electrochem. Soc., 1976, 123, 154–159 CrossRef CAS.
- H. A. H. Alzahrani, J. J. Black, D. Goonetilleke, J. Panchompoo and L. Aldous, Electrochem. Commun., 2015, 58, 76–79 CrossRef CAS.
- H. M. Yau, A. K. Croft and J. B. Harper, Faraday Discuss., 2012, 154, 365–371 RSC.
- L. Aldous, A. Khan, M. M. Hossain and C. Zhao, Catalysis in Ionic Liquids: From Catalyst Synthesis to Application, The Royal Society of Chemistry, 2014, pp. 433–473 Search PubMed.
- (a) E. I. Rogers, D. S. Silvester, D. L. Poole, L. Aldous, C. Hardacre and R. G. Compton, J. Phys. Chem. C, 2008, 112, 2729–2735 CrossRef CAS; (b) C. P. Fu, L. Aldous, E. J. F. Dickinson, N. S. A. Manan and R. G. Compton, ChemPhysChem, 2011, 12, 1708–1713 CrossRef CAS PubMed; (c) C. P. Fu, L. Aldous, E. J. F. Dickinson, N. S. A. Manan and R. G. Compton, Chem. Commun., 2011, 47, 7083–7085 RSC; (d) C. P. Fu, L. Aldous, E. J. F. Dickinson, N. S. A. Manan and R. G. Compton, New J. Chem., 2012, 36, 774–780 RSC.
- (a) E. I. Rogers, D. S. Silvester, L. Aldous, C. Hardacre and R. G. Compton, J. Phys. Chem. C, 2008, 112, 6551–6557 CrossRef CAS; (b) E. I. Rogers, I. Streeter, L. Aldous, C. Hardacre and R. G. Compton, J. Phys. Chem. C, 2008, 112, 10976–10981 CrossRef CAS.
- For example; (a) B. Gélinas and J. C. Forgie, and D. Rochefort, J. Electrochem. Soc., 2014, 161, H161–H165 CrossRef; (b) B. Gélinas and D. Rochefort, Electrochim. Acta, 2015, 162, 36–44 CrossRef; (c) Y. Funasako, T. Mochida, T. Inagaki, T. Sakurai, H. Ohta, K. Furukawa and T. Nakamura, Chem. Commun., 2011, 47, 4475–4477 RSC; (d) T. Inagaki and T. Mochida, Chem. Lett., 2010, 39, 572–573 CrossRef CAS.
- C.-T. Li, C.-P. Lee, C.-T. Lee, S.-R. Li, S.-S. Sun and K.-C. Ho, ChemSusChem, 2015, 8, 1244–1253 CrossRef CAS PubMed.
- (a) H. Grimes and S. R. Logan, Inorg. Chim. Acta, 1980, 45, L223–L224 CrossRef CAS; (b) S. R. Logan and M. R. Welsh, Z. Phys. Chem., 1986, 148, 215–220 CrossRef CAS; (c) S. R. Logan and M. R. Welsh, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 1259–1265 RSC; (d) H. M. A. Salman, M. R. Mahmoud, M. H. M. Abou-El-Wafa, U. M. Rabie and R. H. Crabtree, Inorg. Chem. Commun., 2004, 7, 1209–1212 CrossRef CAS; (e) J. R. B. Hanna and S. R. Logan, J. Photochem., 1979, 10, 267–271 CrossRef CAS; (f) E. W. Neuse and M. S. Loonat, J. Organomet. Chem., 1985, 286, 329–341 CrossRef CAS.
- J. P. Hurvois and C. Moinet, J. Organomet. Chem., 2005, 690, 1829–1839 CrossRef CAS.
- T. Migita, N. Tachikawa, Y. Katayama and T. Miura, Electrochemistry, 2009, 77, 639–641 CrossRef CAS.
- A. Gunawan, H. C. Li, C. H. Lin, D. A. Buttry, V. Mujica, R. A. Taylor, R. S. Prasher and P. E. Phelan, Int. J. Heat Mass Transfer, 2014, 78, 423–434 CrossRef CAS.
- M. J. A. Shiddiky, A. A. J. Torriero, C. Zhao, I. Burgar, G. Kennedy and A. M. Bond, J. Am. Chem. Soc., 2009, 131, 7976–7989 CrossRef CAS PubMed.
- K. Kaya, N. Mikami, Y. Udagawa and M. Ito, Chem. Phys. Lett., 1972, 16, 151–153 CrossRef CAS.
- R. E. Rundle, J. F. Foster and R. R. Baldwin, J. Am. Chem. Soc., 1944, 66, 2116–2120 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, raw data and determination of the ferrocene/ferrocenium ratios. See DOI: 10.1039/c5cc05889a |
| This journal is © The Royal Society of Chemistry 2016 |