
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the reaction mechanism of redox transmetallation of elemental Yb with Hg(C6F5)2 and subsequent reactivity of Yb(C6F5)2 with pyrazole: a DFT investigation†
Julia
Lefèvre
a,
Glen B.
Deacon
b,
Peter C.
Junk
c and
Laurent
Maron
*a
aUniversité de Toulouse et CNRS INSA, UPS, CNRS, UMR 5215, LPCNO, 135 avenue de Rangueil, 31077 Toulouse, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
bSchool of Chemistry, Monash University, Clayton Vic 3800, Australia. E-mail: glen.deacon@monash.edu
cCollege of Science, Technology & Engineering, James Cook University, Townsville, Qld 4811, Australia. E-mail: peter.junk@jcu.edu.au
First published on 17th August 2015
Abstract
DFT investigations of the redox transmetallation reaction of the diorganomercurial (Hg(C6F5)2) with Yb metal, yielding Yb(C6F5)2, allowed us to define a very low energy reaction mechanism. This involves formation of a metal–metal bonded, formally YbI–HgI, intermediate valence complex, (C6F5)Yb–Hg(C6F5). The subsequent reactivity of the divalent ytterbium complex with pyrazole was also computationally investigated, indicating that σ-bond metathesis occurs at divalent ytterbium.
The synthesis of compounds with unsupported lanthanoid-metal bonds (main group (MGM) or transition metal (TM)) has been a challenge, perhaps partly because the 4f orbitals are embedded and are shielded by the 5s25p6 orbitals,1 and partly owing to the intrinsic difficulty of binding together two electropositive elements. Nevertheless recent synthetic ingenuity has enabled a flowering of initially Ln–MGM1,2 and then Ln–TM3 bonded compounds with no supporting donor/bridging atoms. An unsupported Ln–M bond was initially proposed4 in intermediates, namely Yb+–Hg−(C6F5)2 and C6F5Yb–HgC6F5, in the first redox-transmetallation synthesis of an organolanthanoid4,5 (1, and see structure6 of isolated complex [Yb(C6F5)2(thf)4]).
 | (1) |
 | (2) |
Also studied is the mechanism of the cleavage of Yb(C6F5)2 with pyrazole (pzH) (3). Besides being a general model for the protolysis of Yb(C6F5)2 with phenols,10 cyclopentadienes,11 and substituted pyrazoles,12 reactions (1) and (3) or analogues are considered steps in the synthetically valuable redox transmetallation/protolysis (RTP) reaction (4),13 which is an excellent route to both divalent and trivalent cyclopentadienyls,13 aryloxides,14,10a pyrazolates,12 formamidinates15etc. Reaction (1) is considered the first step and reaction (3) the final step for n = 2, whilst reaction (1) is the first step and reaction (3) an intermediate step for n = 3. Thus, the calculations shed light not just specifically on the mechanism of (1) and (3) but also on the general RTP synthesis (4).12–15
| Yb(C6F5)2 + 2pzH → Yb(pz)2 + 2C6F5H | (3) |
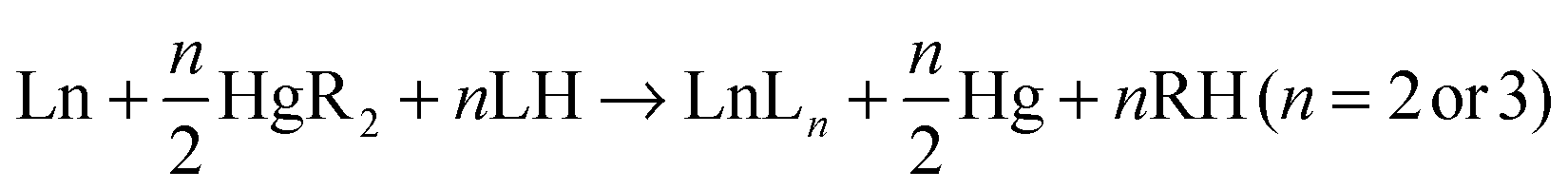 | (4) |
 | ||
| Scheme 1 Computed reaction profile (kcal mol−1) for the redox transmetallation reaction. The F atoms are omitted for clarity in the scheme. | ||
In a first step, the coordination of the ytterbium metal to the mercury center is observed through TS1 with a marginal activation barrier (0.9 kcal mol−1). This coordination compound (Int2) is slightly stabilized (1.8 kcal mol−1) with respect to the separated reactants. It is noteworthy that in Int2, a donor–acceptor interaction between Yb and Hg (donation from the 6s orbital of Yb into the 7p orbital of Hg and a Wiberg bond index of 0.35) is found. Analysis of the NPA charges indicates that the oxidation states of Hg and Yb are both intermediate between 0 and +II (Hg: decrease of the charge from 0.96 to 0.75 and Yb: increase of the charge from 0.0 to 0.24). From Int2, the compound undergoes a migratory insertion of Yb into a C6F5–Hg bond viaTS2. The barrier to this migration is small (4.0 kcal mol−1), indicating a facile process. Interestingly, at the TS, NBO analysis indicates the formation of a covalent bond between Hg and Yb (Wiberg bond index of 0.82) that concomitantly induces a drastic change of the charges (and therefore of the oxidation states) of the two metals (Hg: decrease of the charge from 0.75 to 0.22 and Yb: increase of the charge from 0.24 to 0.96). TS2 yields intermediate Int3 exhibiting an unsupported Yb–Hg interaction (bond found at the second order donor–acceptor NBO and Wiberg bond index of 0.58) with one C6F5 ligand having been fully transferred to Yb. Interestingly, in this intermediate, the Hg–Yb bond is strongly polarized toward Hg as reflected by the NBO analysis (donation from the 7s of Hg to an hybrid d/p of Yb) and the NPA charges (Hg: decrease from 0.22 to 0.1 and Yb: increase from 0.96 to 1.25). By comparison, with the other NPA charges, this complex is of intermediate spin.18 Finally, the second ligand transfer occurs through TS3 with a low activation barrier (6.9 kcal mol−1) yielding an highly thermodynamically stable Hg adduct to Yb(C6F5)2. NBO analysis of the latter reveals at the second order a donor–acceptor interaction between Hg and Yb (donation from 7s of Hg to a d/p hybrid of Yb) with strength computed to be 7.6 kcal mol−1. Other reaction pathways involving for instance Single Electron Transfer, direct double migration of the C6F5 ligand were investigated but either failed to converge (double transfer), were leading to the same profile as the one reported here (Single Electron Transfer) or leads to other energy profiles (ionic dissociation). There might be other plausible pathways but the one reported here is the lowest energy one that was found.
The subsequent reactivity of the divalent ytterbium complex with a nitrogen acid (pyrazole in this case) has then been investigated using the same theoretical approach (Scheme 2).
 | ||
| Scheme 2 Computed reaction profile of the reactivity of the divalent ytterbium complex with pyrazole (kcal mol−1). L stands for pyrazolate. The F atoms and pyrazole/pyrazolate double bonds are omitted for clarity in the scheme. | ||
The “ligand exchange” between Yb(C6F5)2 and the pyrazole is predicted to be thermodynamically favorable (exothermic by 27.9 kcal mol−1) and can occur via a dissociative pathway (energetic cost of only 7.6 kcal mol−1). From the pyrazolate adduct to the divalent ytterbium complex, two consecutive N–H activations can easily occur with moderate activation barriers (7.0 and 13.0 kcal mol−1). These two N–H activations are classical σ-bond metatheses, where N, H and Cipso are almost aligned,17 and can be viewed as proton transfer between the two ligands. In line with the Hard and Soft Acid-Base (HSAB) principle, the formation of a Yb–N bond is favored over the Yb–C one, explaining the exothermicity of the reaction.
In this study, the redox transmetallation reaction of Hg(C6F5)2 with metallic Yb has been computed at the DFT level. The reaction is found to be kinetically and thermodynamically favourable. The formation of a transient complex bearing a Hg–Yb bond is also predicted, that rapidly evolves to the final divalent ytterbium complex. The latter can then react with a proton donor (pyrazole in this study) through σ-bond metathesis. This reaction is also kinetically and thermodynamically favourable and indicates the propensity of divalent ytterbium to react without change of oxidation state. The challenge now arises for us to isolate an intermediate involving a Yb–Hg bonded species.
We thank the Alexander von Humboldt Foundation for a fellowship to L.M. and the Australian Research Council for Grant DP130100152.
Notes and references
- M. T. Gamer, P. W. Roesky, S. N. Konchenko, P. Nava and R. Ahlrichs, Angew. Chem., Int. Ed., 2006, 45, 4447 CrossRef CAS.
- Recent example (a) S. T. Liddle, P. Mills, B. M. Gardner, C. Jones and W. D. Woods, Inorg. Chem., 2009, 48, 3250 Search PubMed; (b) I. L. Fedushkin, A. N. Lukoyanov, A. N. Tishkina, M. O. Maslov, S. U. Ketkov and H. Hummert, Organometallics, 2011, 30, 3628 CrossRef CAS; (c) C. Jones, A. Stasch and W. Woodal, Chem. Commun., 2009, 113 RSC; (d) S. G. Minasian, J. L. Krinsky, J. D. Rinehart, R. Copping, T. Tyliszczak, M. Janousch, D. K. Shuh and J. Arnold, J. Am. Chem. Soc., 2009, 131, 13767 CrossRef CAS PubMed; (e) M. Wiecko and P. W. Roesky, Organometallics, 2007, 26, 4846 CrossRef CAS; (f) T. Sanden, M. T. Gamer, A. A. Fagin, V. A. Chudakova, S. N. Konchenko, I. L. Fedushkin and P. W. Roesky, Organometallics, 2012, 31, 4331 CrossRef CAS; (g) K. Zeckert, S. Zahn and B. Kirchner, Chem. Commun., 2010, 46, 2638 RSC; (h) K. Zeckert, J. Kriebel, R. Kirmse, M. Weiss and R. Denecke, Chem. – Eur. J., 2013, 19, 7718 CrossRef CAS PubMed; (i) P. L. Arnold, S. T. Liddle, J. McMaster, C. Jones and D. P. Mills, J. Am. Chem. Soc., 2007, 129, 5360 CrossRef CAS PubMed.
- For example: (a) I. P. Beletskaya, A. Z. Voskoboynikov, E. B. Choklanova, N. I. Kirillova, A. K. Shestakova, I. N. Parshina, A. I. Gusev and G. K.-I. Magomedov, J. Am. Chem. Soc., 1993, 115, 3156 CrossRef CAS; (b) M. Butovskii, O. Tok, F. Wagner and R. Kempe, Angew. Chem., Int. Ed., 2008, 47, 6469 CrossRef CAS; (c) M. V. Butovskii, C. Doring, V. Bezugly, F. R. Wagner, Y. Grin and R. Kempe, Nat. Chem., 2010, 2, 741 CrossRef CAS; (d) C. Doring, A.-M. Dietel, M. V. Butovskii, V. Bezugly, F. R. Wagner and R. Kempe, Chem. – Eur. J., 2010, 16, 10679 CrossRef PubMed; (e) M. P. Blake, N. Kaltsoyannis and P. Mountford, Chem. Commun., 2013, 49, 331 RSC; (f) P. L. Arnold, J. McMaster and S. T. Liddle, Chem. Commun., 2009, 818 RSC; (g) M. V. Butovski, B. Oelkers, T. Bauer, J. M. Bakker, V. Bezugly, F. F. Wagner and R. Kempe, Chem. – Eur. J., 2014, 20, 2804 CrossRef.
- G. B. Deacon, W. D. Raverty and D. G. Vince, J. Organomet. Chem., 1977, 135, 103 CrossRef CAS.
- G. B. Deacon and D. G. Vince, J. Organomet. Chem., 1976, 112, C1 CrossRef CAS.
- G. B. Deacon and C. M. Forsyth, Organometallics, 2003, 22, 1349 CrossRef CAS.
- G. Z. Suleimanov, L. F. Rybakova, Y. a. A. Nuriev, T. K. h. Kurbanov and I. P. Beletskaya, Izv. Akad. Nauk SSSR, Ser. Khim., 1982, 1983, 32, 190 Search PubMed.
- M. N. Bochkarev, L. N. Zakharova and G. S. Kalinina, Organoderivatives of Rare Earth Elements, Kluwer Academic, Dordrecht, 1995 Search PubMed.
- A. E. Godard and D. Goddard, Organometallic Compounds, in A Textbook of Inorganic Chemistry, ed. J. N. Friend, Griffen, London, 1928, vol. XI, part 1, pp. 32–36, 74–77 Search PubMed.
- (a) G. B. Deacon, P. B. Hitchcock, S. A. Holmes, M. F. Lappert, P. MacKinnon and R. H. Newnham, J. Chem. Soc., Chem. Commun., 1989, 935 RSC; (b) G. B. Deacon, T. Feng, P. MacKinnon, R. H. Newnham, S. Nickel, B. W. Skelton and A. H. White, Aust. J. Chem., 1993, 46, 387 CrossRef CAS.
- G. B. Deacon and R. H. Newnham, Aust. J. Chem., 1985, 38, 757 Search PubMed.
- G. B. Deacon, E. E. Delbridge, B. W. Skelton and A. H. White, Eur. J. Inorg. Chem., 1999, 751 CrossRef CAS.
- G. B. Deacon, C. M. Forsyth and S. Nickel, J. Organomet. Chem., 2002, 647, 50 CrossRef CAS.
- G. B. Deacon, G. D. Fallon, C. M. Forsyth, S. C. Harris, P. C. Junk, B. W. Skelton and A. H. White, Dalton Trans., 2006, 802 RSC.
- (a) M. L. Cole, G. B. Deacon, C. M. Forsyth, P. C. Junk, K. Konstas and J. Wang, Chem. – Eur. J., 2007, 13, 8092 CrossRef CAS PubMed; (b) M. L. Cole, G. B. Deacon, C. M. Forsyth, P. C. Junk, K. Konstas, J. Wang, H. Bittig and D. Werner, Chem. – Eur. J., 2013, 19, 1410 CrossRef CAS PubMed.
- (a) L. Castro, C. E. Kefalidis, D. McKay, S. Essafi, L. Perrin and L. Maron, Dalton Trans., 2014, 43, 12124 RSC; (b) L. Maron, L. Perrin, L. Castro, A. Yahia and C. E. Kefalidis, in Computational Methods in Lanthanide and Actinide Chemistry, ed M. Dolg, Wiley, 2015, p. 343 Search PubMed.
- L. Maron and O. Eisenstein, J. Phys. Chem. A, 2000, 104, 7140 CrossRef CAS.
- V. Grignard - Nobel Lecture: The Use of Organomagnesium Compounds in Preparative Organic Chemistry”. http://Nobelprize.org. Nobel Media AB 2014.
Footnote |
| † Electronic supplementary information (ESI) available: Details of DFT calculations. See DOI: 10.1039/c5cc05439g |
| This journal is © The Royal Society of Chemistry 2015 |