
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iridium-catalyzed asymmetric cyclization of alkenoic acids leading to γ-lactones†
Midori
Nagamoto
and
Takahiro
Nishimura
*
Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo, Kyoto 606-8502, Japan. E-mail: tnishi@kuchem.kyoto-u.ac.jp
First published on 17th July 2015
Abstract
Asymmetric cyclization of alkenoic acids was realized by the use of an iridium/chiral bisphosphine catalyst, giving high yields of the corresponding γ-lactones with good enantioselectivity.
Carboxylic acid esters are one of the most ubiquitous and important compounds, and are also of great value as synthetic intermediates leading to the formation of alcohols via saponification or reduction. Among a variety of approaches to the esters, the catalytic addition of carboxylic acids to C–C unsaturated bonds provides highly atom-efficient and straightforward methodologies.1 Transition metal-catalyzed addition of carboxylic acids was pioneered by Shvo and Rotem, who reported the Ru-catalyzed addition of carboxylic acids to alkynes,2a,b and there have been many successful examples of the addition to alkynes2 and allenes.3,4 Recently, the addition of carboxylic acids to unactivated alkenes has also been achieved by the use of Ru,5 Fe,6 Au,7 and other metal catalysts.8
Asymmetric addition of oxygen nucleophiles to unsaturated bonds is still a challenging objective in organic chemistry. The successful examples of asymmetric addition have been limited to reactive unsaturated bonds such as allenes.9–11 The research groups of Toste9 and Widenfoefer10 reported Au-catalyzed asymmetric intramolecular hydroalkoxylation of allenes independently. In this context, Breit and co-workers recently reported that a Rh/chiral bisphophine complex can efficiently catalyze the asymmetric intermolecular addition of carboxylic acids to allenes to give allylic esters with high enantioselectivity.12 Meanwhile, there have been few reports on the asymmetric addition of oxygen-nucleophiles to simple alkenes. Hartwig and co-workers reported Ir-catalyzed intermolecular hydroalkoxylation of alkenes with a modest ee.13 Hintermann and co-workers recently reported an asymmetric intramolecular hydroalkoxylation of allylphenols catalyzed by a Ti complex.14
An inherent problem in the asymmetric addition of carboxylic acids to alkenes is a non-asymmetric background reaction catalyzed by a strong Brønsted acid, which has been reported to be sometimes formed by use of metal triflates or cationic metal catalysts.15 We found that a neutral Ir complex can catalyze the intramolecular addition of a carboxylic acid to an alkene. Here we report the iridium-catalyzed asymmetric cyclization of alkenic acids to give the corresponding γ-lactones in high yields with good enantioselectivity.
Treatment of 2,2-diphenyl-4-pentenoic acid (1a) in the presence of [IrCl(coe)2]2 (5 mol% of Ir) and (R)-binap16 in 1,4-dioxane at 80 °C for 20 h gave lactone 2a in 8% yield with 13% ee (Table 1, entry 1). The use of (R)-H8-binap17 was also less effective in both the yield and the enantioselectivity (entry 2). (S)-Segphos,18 (R)-difluorophos,19 and (R)-DTBM-segphos18 displayed better enantioselectivity than (R)-binap (entries 3–5), where the use of (R)-DTBM-segphos gave 2a with 58% ee (entry 5). The use of a non-polar solvent such as toluene resulted in a low yield and enantioselectivity (entry 6). A significant improvement of the catalytic activity and enantioselectivity was observed by the use of N-methylpyrrolidone (NMP) as a solvent, giving the lactone in 39% yield with 80% ee (entry 7). The reaction at higher temperature (100 °C) in NMP gave 2a in 95% yield without a decrease of the enantioselectivity (81% ee, entry 8). An Ir complex [IrCl(coe)2]2 without the phosphine ligand displayed a very low catalytic activity (entry 9). A rhodium catalyst did not promote the present cyclization (entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Yieldb (%) | eec (%) |
| a Reaction conditions: carboxylic acid 1a (0.10 mmol), [IrCl(coe)2]2 (5 mol% of Ir) and ligand (5 mol%) in solvent (0.40 mL) at 80 °C for 20 h. b Determined by 1H NMR analysis. c Determined by chiral HPLC analysis. d At 100 °C. e [RhCl(cod)]2 was used instead of [IrCl(coe)2]2. NMP = N-methyl-2-pyrrolidone. | ||||
| 1 | (R)-Binap | 1,4-Dioxane | 8 | 13 |
| 2 | (R)-H8-binap | 1,4-Dioxane | 14 | 10 |
| 3 | (S)-Segphos | 1,4-Dioxane | 12 | 23 |
| 4 | (R)-Difluorophos | 1,4-Dioxane | 11 | 25 |
| 5 | (R)-DTBM-segphos | 1,4-Dioxane | 13 | 58 |
| 6 | (R)-DTBM-segphos | Toluene | 5 | 29 |
| 7 | (R)-DTBM-segphos | NMP | 39 | 80 |
| 8d | (R)-DTBM-segphos | NMP | 95 | 81 |
| 9 | None | NMP | 4 | — |
| 10e | (R)-Binap | NMP | 0 | — |
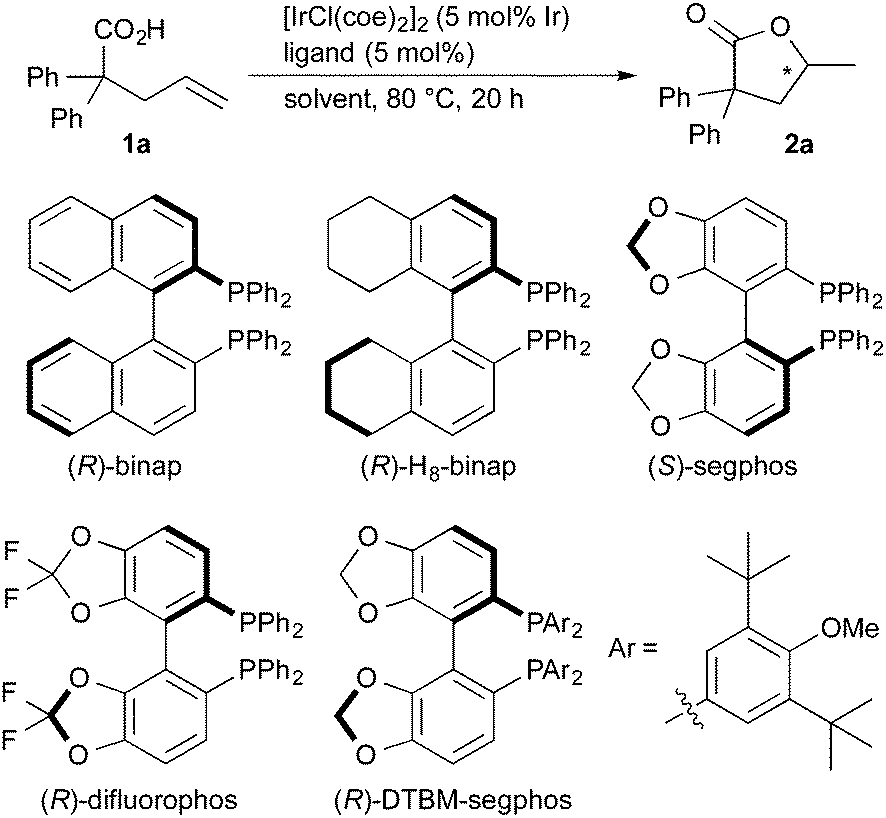
The results obtained for the iridium-catalyzed asymmetric cyclization of alkenoic acids are summarized in Table 2. Several 2,2-disubstituted 4-pentenoic acids 1 underwent the cyclization to give the corresponding lactones 2 (entries 1–7). The reaction of 2,2-diaryl-4-pentenoic acids 1b and 1c, having electron-donating substituents on the aromatic rings, proceeded to give 2b and 2c in high yields with 85 and 83% ee, respectively (entries 2 and 3). The reaction of 1d and 1e, having electron-withdrawing fluoro and chloro groups, gave 2d and 2e with modest enantioselectivity, 70 and 61% ee, respectively (entries 4 and 5). The modest yield (53%) of 2e is due to the loss of 1e by decarboxylation, which was proven to proceed without the iridium catalyst under the reaction conditions: heating of 1e in NMP at 100 °C for 20 h gave 4,4-di(4-chlorophenyl)-1-butene in 56% yield (66% conversion of 1e). 2,2-Dialkyl-4-pentenoic acids 1f and 1g are also good substrates to give corresponding lactones 2f and 2g in good yields with 86 and 89% ee, respectively (entries 6 and 7). The substituents at the 2-position of carboxylic acids 1 are essential for the present reaction: 2-pentenioc acid did not undergo the cyclization.20 On the other hand, the reaction of 2-vinylbenzoic acid 1h proceeded at 120 °C to give 2h with a moderate enantioselectivity (entry 8).21
 | (1) |
 | (2) |
|
|
||||
|---|---|---|---|---|
| Entry | R | Yieldb (%) | eec (%) | |
| a Reaction conditions: carboxylic acid 1 (0.20 mmol), [IrCl(coe)2]2 (5 mol% Ir) and (R)-DTBM-segphos in NMP (0.80 mL) at 100 °C for 20 h. b Isolated yield. c Determined by chiral HPLC analysis. d For 48 h. e At 120 °C. | ||||
| 1 | Ph | 1a | 92 (2a) | 81 |
| 2 | 4-MeC6H4 | 1b | 98 (2b) | 85 |
| 3 | 4-MeOC6H4 | 1c | 96 (2c) | 83 |
| 4 | 4-FC6H4 | 1d | 86 (2d) | 70 |
| 5 | 4-ClC6H4 | 1e | 53 (2e) | 61 |
| 6d | Benzyl | 1f | 87 (2f) | 86 |
| 7d | n-Hexyl | 1g | 74 (2g) | 89 |
| 8e | — | 1h | 57 (2h) | 49 |
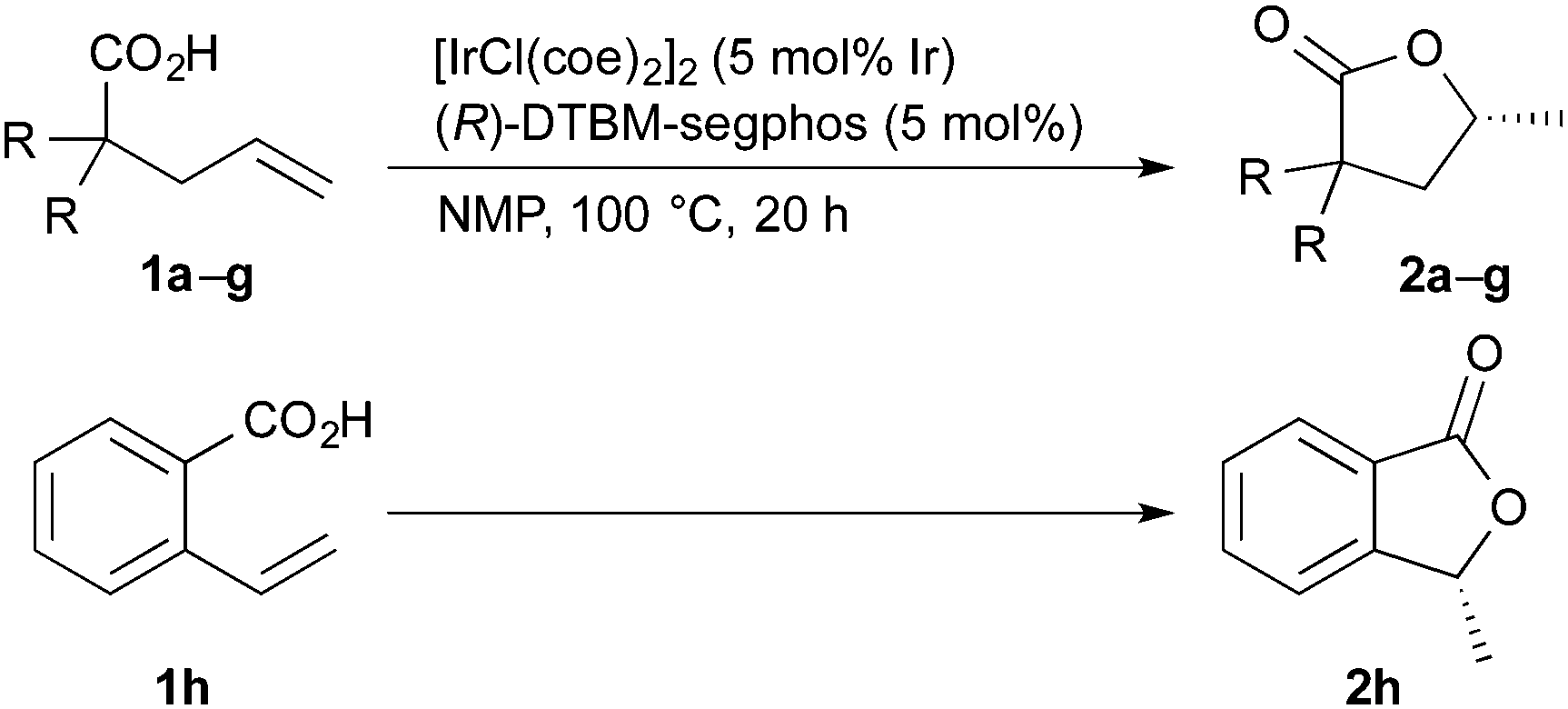
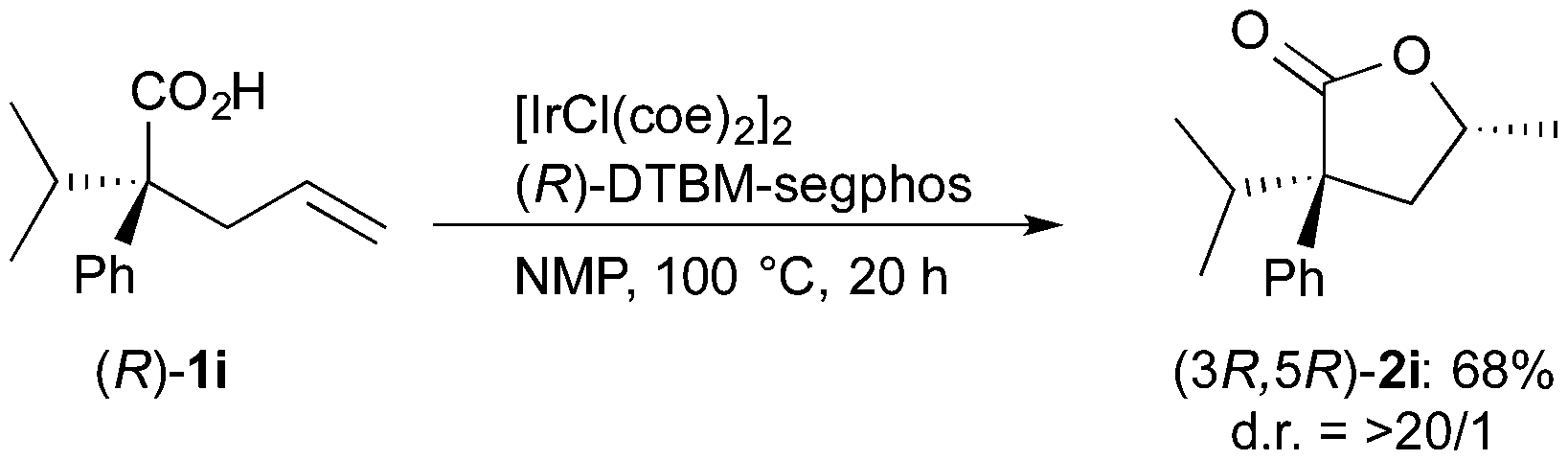
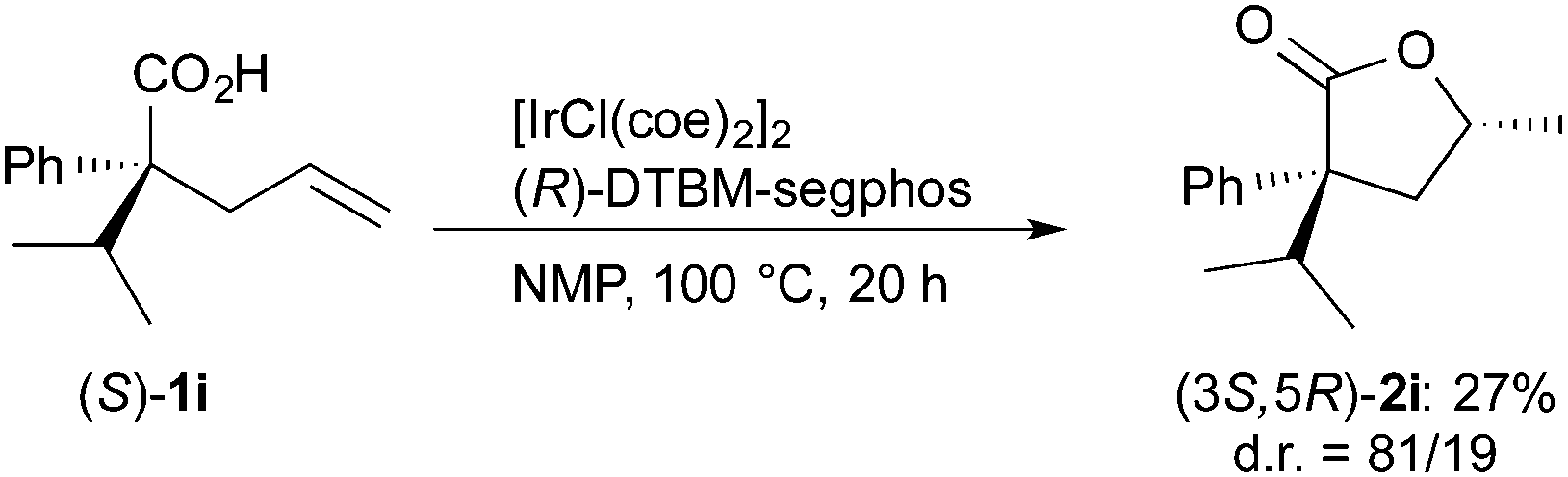
The stereochemistry of the lactone formed in the present catalytic conditions was estimated by the reaction of enantiomerically pure carboxylic acids 1i. The reaction of (R)-1i in the presence of the Ir/(R)-DTBM-segphos catalyst gave lactone 2i in 68% yield with very high diastereoselectivity (eqn (1)). The absolute configuration of the lactone 2i was determined to be 3R,5R, which was assigned by comparison of the optical rotation ([α]D = −27, c 0.74 in CHCl3) with the reported one ([α]D = −45.3, c 1.17 in CHCl3 for (3R,5R)-2i).22 On the other hand, a lower diastereoselectivity 81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 was observed in the reaction of (S)-1i (eqn (2)), indicating that the face-selectivity of the cyclization is influenced by the substituents at the α-position of the carboxylic acids 1.
:19 was observed in the reaction of (S)-1i (eqn (2)), indicating that the face-selectivity of the cyclization is influenced by the substituents at the α-position of the carboxylic acids 1.
 | (3) |
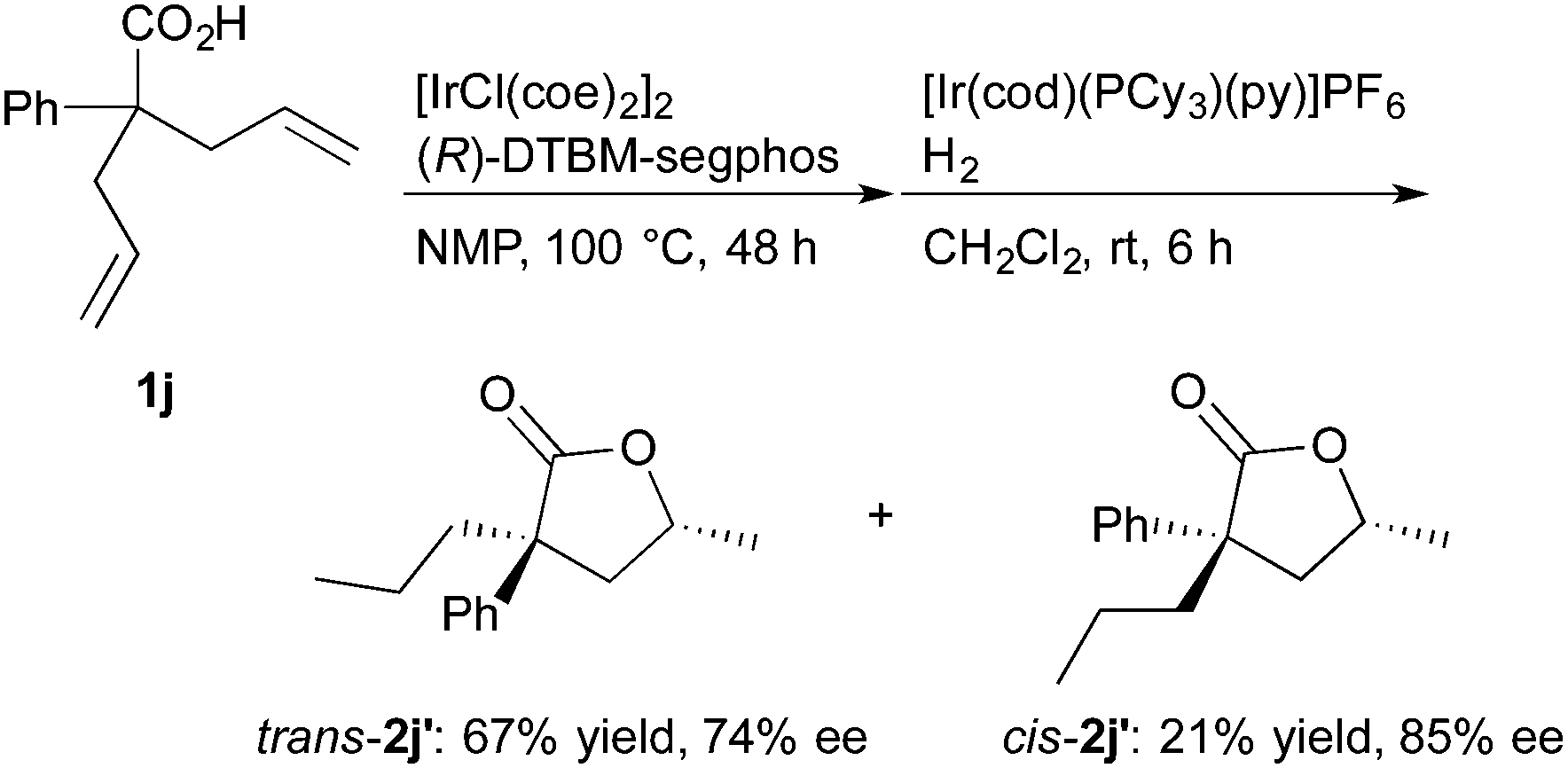
The reaction of 2,2-diallylphenylacetic acid (1j) proceeded well to give the corresponding lactones in good yields, where the lactones contained double bond isomers and they were hydrogenated in the presence of [Ir(cod)(PCy3)(py)]PF623 (Cy = cyclohexyl, py = pyridine, eqn (3)). The saturated lactones trans-2j′ and cis-2j′ were formed in moderate diastereoselectivity and good enantioselectivity.
Scheme 1 shows three possible reaction pathways for the Ir-catalyzed cyclization. One involves the oxidative addition of the carboxylic acid 1a to the Ir(I) giving a hydridoiridium(III) species, and a sequential alkene insertion into the Ir(III) leads to the formation of lactone 2avia reductive elimination (Scheme 1a). Another reaction pathway is associated with the formation of an Ir(I) carboxylate species (Scheme 1b). In consideration of a weak basicity of NMP, the Ir(I) carboxylate species could be formed by deprotonation and the species undergoes the alkene insertion. Scheme 1c shows the other pathway initiated by an electrophilic activation of the alkene moiety with the Ir(I) species, where the subsequent attack of the carboxyl group to the alkene forms the C–O bond. Mashima and co-workers reported the synthesis of hydridoiridium(III) carboxylate complexes via oxidative addition of carboxylic acids to an Ir(I)/binap complex.24 Krische and co-workers reported an iridium-catalyzed addition of carboxylic acids to allenes,3c where it is proposed that oxidative addition is the initial step of the reaction. To gain some insight into the mechanism, a stoichiometric reaction of 1a with [IrCl(coe)2]2 and (R)-DTBM-segphos in benzene-d6 was conducted. Treatment of [IrCl(coe)2]2, (R)-DTBM-segphos, and carboxylic acid 1a in benzene-d6 at room temperature for 24 h brought about the formation of hydridoiridium complexes as a mixture of two isomers (73:27). The major isomer showed a virtual triplet at −27.1 ppm (JP–H = 22 Hz) in the 1H NMR analysis, which was tentatively assigned to be a hydride at a cis-position to two phosphorous atoms.25 The result indicates that the reaction pathway (a) initiated by the oxidative addition of the carboxylic acid is plausible in the present cyclization.
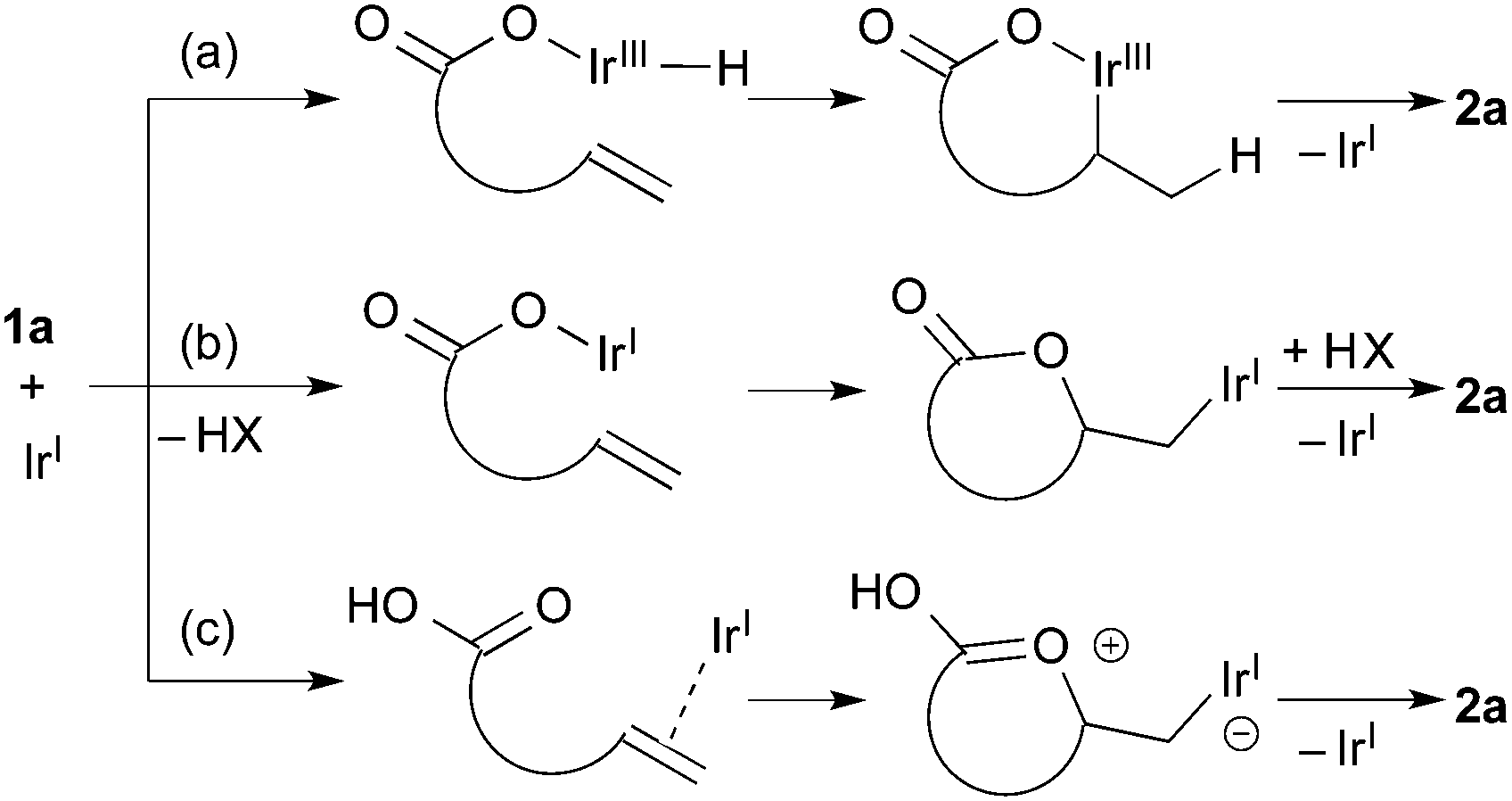 | ||
| Scheme 1 Possible reaction pathways. | ||
The possible intermediacy of the iridium(I) carboxylate species was also investigated by the use of a hydroxoiridium(I) complex as a catalyst precursor. The reaction of 1a was conducted in the presence of [Ir(OH)(cod)]2 and (R)-DTBM-segphos, which is expected to react with 1a to form the iridium(I) carboxylate. The reaction gave the lactone in 20% yield accompanied by decarboxylation products in 62% yield as a mixture of the double bond isomers.26 The result indicates that the iridium(I) carboxylate is not likely to be the intermediate in the present reaction, because the formation of such decarboxylation products was not observed in the reaction of 1a catalyzed by the IrCl/(R)-DTBM-segphos complex.
Determining the stereochemistry of the addition using deuterated carboxylic acids would be helpful in distinguishing pathway (c) from others; pathway (c) leads to an anti-addition product while others lead to a syn-addition product. Unfortunately, however, the reactions of carboxylic acids containing internal alkenes were unsuccessful, and thus, the pathway (c) could not be excluded at this stage.26
The results of deuterium-labeling experiments are shown in eqn (4) and (5). In the reaction of deuterated carboxylic acid 1a-d1, deuterium incorporation into a methyl group of 2a was low (0.15D/3H, eqn (4)). The low content of the deuterium is probably due to an incorporation of hydrogen atoms from solvent NMP via the C–H activation of a methyl group on NMP.27 A 5% of deuterium incorporation at the γ-position was also observed. On the other hand, in the reaction of 1a-d2, which is substituted at the alkene terminus with two deuterium atoms, a significant amount of a migration of deuterium into the γ-position of 2a was observed (0.44D, eqn (5)). A migration of deuterium from the terminal position to the internal one was also detected in a recovered 1a-d2.
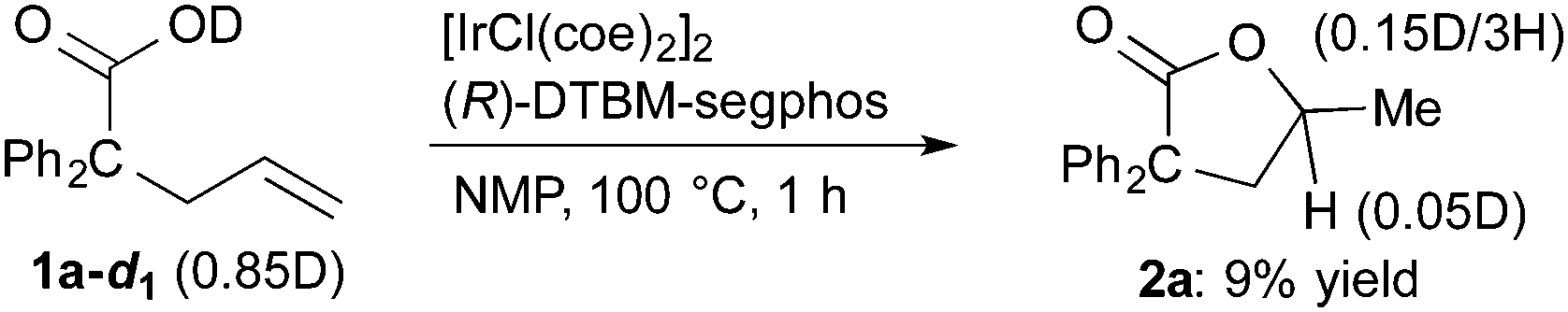 | (4) |
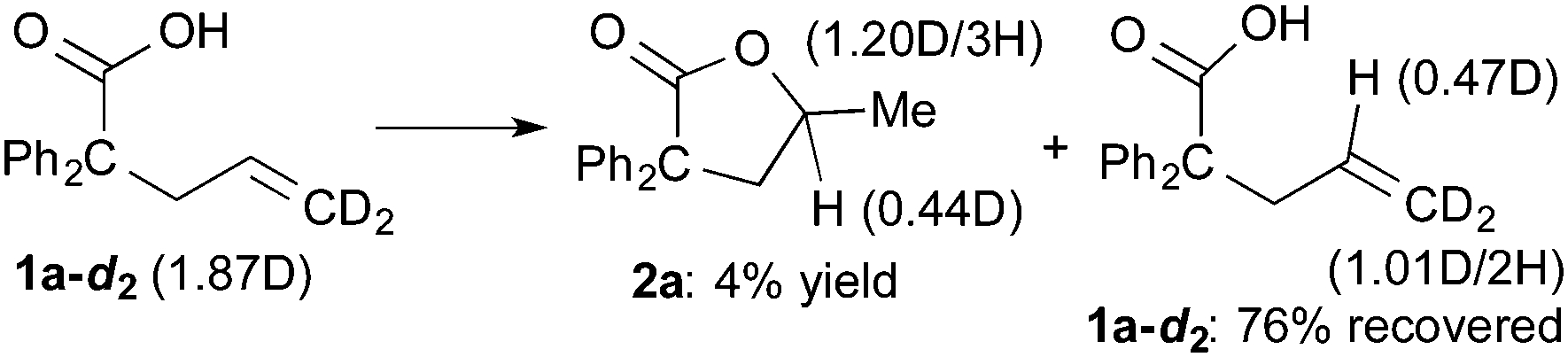 | (5) |
In light of the results of the stoichiometric reaction and deuterium-labeling experiments, the catalytic cycle is postulated as illustrated in Scheme 2. Oxidative addition of O–H bond to Ir(I) forms (carboxylato)iridium(III) hydride B. The alkene insertion into the Ir–H bond in an exo-fashion forms alkyliridium(III) C and the successive reductive elimination gives lactone 2a and regenerates the Ir(I) species. The migration of deuterium observed as shown in eqn (5) can be explained by reversible insertion and β-hydride elimination of intermediate C′, which is formed via the alkene insertion in an endo-fashion.
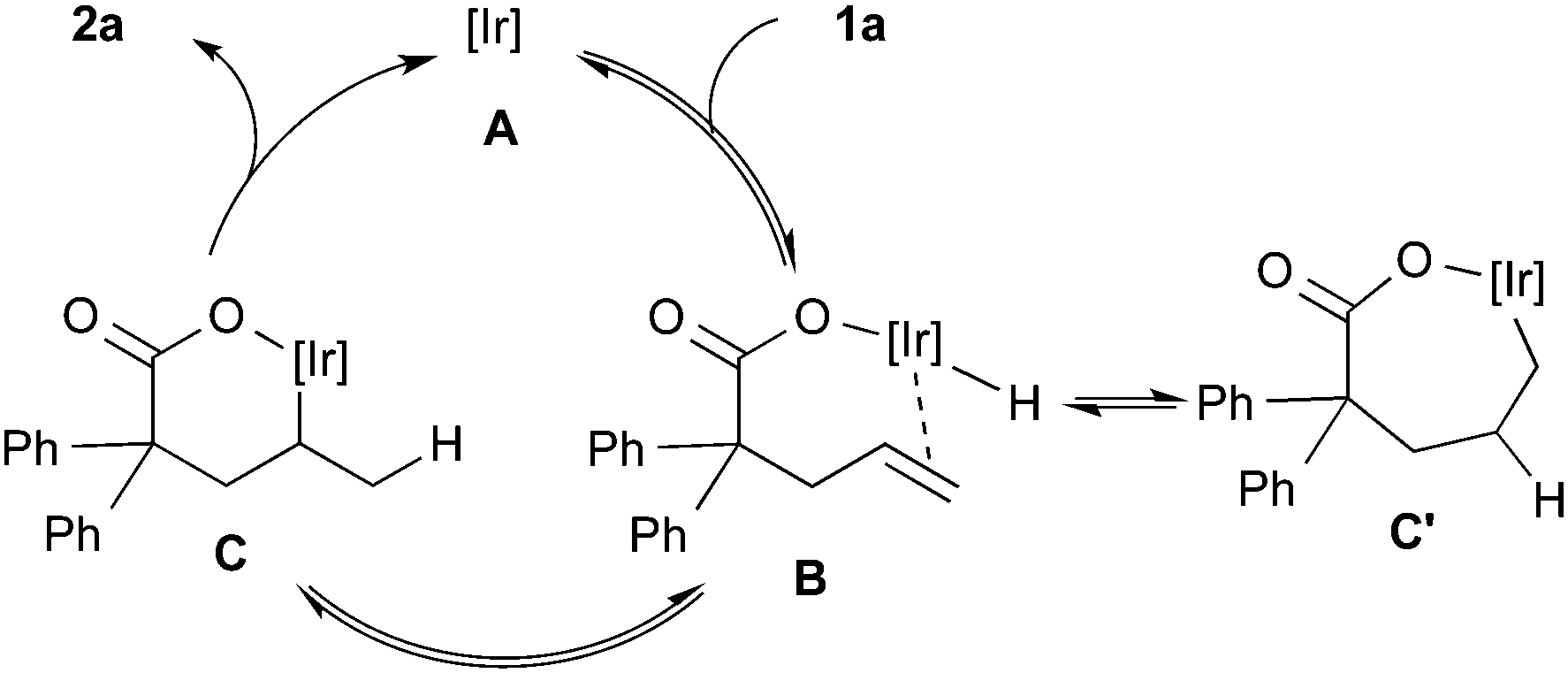 | ||
| Scheme 2 Plausible catalytic cycle. | ||
In summary, we have developed an asymmetric cyclization of alkenoic acids using an Ir/(R)-DTBM-segphos catalyst that gives lactones with good enantioselectivity.
This work was supported by JSPS KAKENHI Grant No. 15H03810. M.N. thanks the JSPS for a Research Fellowship for Young Scientists. We thank Prof. K. Maruoka and Prof. T. Kano (Kyoto University) for HRMS analysis and Takasago International Corporation for the gift of (R)-DTBM-segphos.
Notes and references
- For recent reviews on transition metal-catalyzed addition of carboxylic acids, see: (a) E. M. Barreiro, L. A. Adrio, K. K. Hii and J. B. Brazier, Eur. J. Org. Chem., 2013, 1027 CrossRef CAS PubMed; (b) N. T. Patil, R. D. Kavthe and V. S. Shinde, Tetrahedron, 2012, 68, 8079 CrossRef CAS PubMed; (c) M. P. Munoz, Org. Biomol. Chem., 2012, 10, 3584 RSC; (d) Y. Yamamoto and U. Radhakrishnan, Chem. Soc. Rev., 1999, 28, 199 RSC; (e) C. Bruneau and P. H. Dixneuf, Chem. Commun., 1997, 507 RSC.
- For selected examples of the addition to alkynes, see: (a) M. Rotem and Y. Shvo, Organometallics, 1983, 2, 1689 CrossRef CAS; (b) M. Rotem and Y. Shvo, J. Organomet. Chem., 1993, 448, 189 CrossRef CAS; (c) T. Mitsudo, Y. Hori and Y. Watanabe, J. Org. Chem., 1985, 50, 1566 CrossRef CAS; (d) T. Mitsudo, Y. Hori, Y. Yamakawa and Y. Watanabe, J. Org. Chem., 1987, 52, 2230 CrossRef CAS; (e) H. Doucet, J. Hofer, C. Bruneau and P. H. Dixneuf, J. Chem. Soc., Chem. Commun., 1993, 850 RSC; (f) H. Doucet, B. Martin-Vaca, C. Bruneau and P. H. Dixneuf, J. Org. Chem., 1996, 60, 7247 CrossRef; (g) K. Melis, P. Samulkiewicz, J. Rynkowski and F. Verpoort, Tetrahedron Lett., 2002, 43, 2713 CrossRef CAS; (h) H. Nakagawa, Y. Okimoto, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 2003, 44, 103 CrossRef CAS; (i) L. J. Goossen, J. Paetzold and D. Koley, Chem. Commun., 2003, 706 RSC; (j) R. Hua and X. Tian, J. Org. Chem., 2004, 69, 5782 CrossRef CAS PubMed; (k) S. Ye and W. K. Leong, J. Organomet. Chem., 2006, 691, 1117 CrossRef CAS PubMed; (l) F. Nicks, R. Aznar, D. Sainz, G. Muller and A. Demonceau, Eur. J. Org. Chem., 2009, 5020 CrossRef CAS PubMed; (m) C. S. Yi and R. Gao, Organometallics, 2009, 28, 6585 CrossRef CAS PubMed; (n) D.-M. Cui, Q. Meng, J.-Z. Zheng and C. Zhang, Chem. Commun., 2009, 1577 RSC; (o) S. Karabulut, B. Ö. Öztürk and Y. Îmamoglu, J. Organomet. Chem., 2010, 695, 2161 CrossRef CAS PubMed; (p) A. Lumbroso, N. R. Vautravers and B. Breit, Org. Lett., 2010, 12, 5498 CrossRef CAS PubMed; (q) B. C. Chary and S. Kim, J. Org. Chem., 2010, 75, 7928 CrossRef CAS PubMed; (r) V. Cadierno, J. Francos and J. Gimeno, Organometallics, 2011, 30, 852 CrossRef CAS; (s) N. Tsukada, A. Takahashi and Y. Inoue, Tetrahedron Lett., 2011, 52, 248 CrossRef CAS PubMed.
- For selected examples of the addition to allenes, see. (a) M. Al-Masum and Y. Yamamoto, J. Am. Chem. Soc., 1998, 120, 3809 CrossRef CAS; (b) S. Ma, Z. Yu and S. Wu, Tetrahedron, 2001, 57, 1585 CrossRef CAS; (c) I. S. Kim and M. J. Krische, Org. Lett., 2008, 10, 513 CrossRef CAS PubMed.
- (a) W. Zhang, A. R. Haight and M. C. Hsu, Tetrahedron Lett., 2002, 43, 6575 CrossRef CAS; (b) Z. Huo, N. T. Patil, T. Jin, N. K. Pahadi and Y. Yamamoto, Adv. Synth. Catal., 2007, 349, 680 CrossRef CAS PubMed; (c) A. Lumbroso, P. Koschker, N. R. Vautravers and B. Breit, J. Am. Chem. Soc., 2011, 133, 2386 CrossRef CAS PubMed.
- (a) Y. Oe, T. Ohta and Y. Ito, Chem. Commun., 2004, 1620 RSC; (b) T. Ohta, Y. Kataoka, A. Miyoshi, Y. Oe, I. Furukawa and Y. Ito, J. Organomet. Chem., 2007, 692, 671 CrossRef CAS PubMed; (c) Y. Oe, T. Ohta and Y. Ito, Tetrahedron Lett., 2010, 51, 2806 CrossRef CAS PubMed.
- (a) K. Komeyama, Y. Mieno, S. Yukawa, T. Morimoto and K. Takaki, Chem. Lett., 2007, 36, 752 CrossRef CAS; (b) J.-C. Choi, K. Kohno, D. Masuda, H. Yasuda and T. Sakakura, Chem. Commun., 2008, 777 RSC.
- C.-G. Yang and C. He, J. Am. Chem. Soc., 2005, 127, 6966 CrossRef CAS PubMed.
- Cu: (a) J. G. Taylor, N. Whittall and K. K. Hill, Chem. Commun., 2005, 5103 RSC; (b) X. Chaminade, L. Coulombel, S. Olivero and E. Dunach, Eur. J. Org. Chem., 2006, 3554 CrossRef CAS PubMed; (c) L. A. Adrio, L. S. Quek, J. G. Taylor and K. K. Hii, Tetrahedron, 2009, 65, 10334 CrossRef CAS PubMed ; Ag: ; (d) C.-G. Yang, N. W. Reich, Z. Shi and C. He, Org. Lett., 2005, 7, 4553 CrossRef CAS PubMed; (e) L. J. Gooßen, D. M. Ohlmann and M. Dierker, Green Chem., 2010, 12, 197 RSC.
- G. Hamilton, E. J. Kang, M. Mba and F. D. Toste, Science, 2007, 317, 496 CrossRef CAS PubMed.
- Z. Zhang and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2007, 46, 283 CrossRef CAS PubMed.
- (a) K. Aikawa, M. Kojima and K. Mikami, Adv. Synth. Catal., 2010, 352, 3131 CrossRef CAS PubMed; (b) Y. Wang, K. Zheng and R. Hong, J. Am. Chem. Soc., 2012, 134, 4096 CrossRef CAS PubMed; (c) J. L. Arbour, H. Rzepa, S. J. Contreras-Garcia, L. A. Adrio, E. M. Barreiro and K. K. Hii, Chem. – Eur. J., 2012, 18, 11317 CrossRef CAS PubMed; (d) W. Lim, J. Kim and Y. H. Rhee, J. Am. Chem. Soc., 2014, 136, 13618 CrossRef CAS PubMed; (e) T. Kawamoto, S. Hirabayashi, X.-X. Guo, T. Nishimura and T. Hayashi, Chem. Commun., 2009, 3528 RSC.
- (a) P. Koschker, A. Lumbroso and B. Breit, J. Am. Chem. Soc., 2011, 133, 20746 CrossRef CAS PubMed; (b) P. Koschker, M. Kähny and B. Breit, J. Am. Chem. Soc., 2015, 137, 3131 CrossRef CAS PubMed.
- C. S. Sevov and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 9303 CrossRef CAS PubMed.
- J. Schlüter, M. Blazejak, F. Boeck and L. Hintermann, Angew. Chem., Int. Ed., 2015, 54, 4014 CrossRef PubMed.
- (a) R. E. McKinney Brooner and R. A. Widenhoefer, Chem. – Eur. J., 2011, 17, 6170 CrossRef CAS PubMed ; For examples of Brønsted acid catalysis in the presence of metal catalysts, see: ; (b) T. C. Wabnitz, J.-Q. Yu and J. B. Spencer, Chem. – Eur. J., 2004, 10, 484 CAS; (c) D. C. Rosenfeld, S. Shekhar, A. Takemiya, M. Utsunomiya and J. F. Hartwig, Org. Lett., 2006, 8, 4179 CrossRef CAS PubMed; (d) W.-M. Liu, Y. L. Tnay, K. P. Gan, Z.-H. Liu, W. H. Tyan and K. Narasaka, Helv. Chim. Acta, 2012, 95, 1953 CrossRef CAS PubMed; (e) M. J.-L. Tschan, C. M. Thomas, H. Strub and J.-F. Carpentier, Adv. Synth. Catal., 2009, 351, 2496 CrossRef CAS PubMed; (f) E. Szuromi and P. R. Sharp, Organometallics, 2006, 25, 558 CrossRef CAS; (g) R. E. M. Brooner, B. D. Robertson and R. A. Widenhoefer, Organometallics, 2014, 33, 6466 CrossRef CAS; (h) R. Dumeunier and I. E. Markó, Tetrahedron Lett., 2004, 45, 825 CrossRef CAS PubMed.
- H. Takaya, K. Mashima, K. Koyano, M. Yagi, H. Kumobayashi, T. Taketomi, S. Akutagawa and R. Noyori, J. Org. Chem., 1986, 51, 629 CrossRef CAS.
- X. Zhang, K. Mashima, K. Koyano, N. Sayo, H. Kumobayashi, S. Akutagawa and H. Takaya, Tetrahedron Lett., 1991, 32, 7283 CrossRef CAS.
- T. Saito, T. Yokozawa, T. Ishizaki, T. Moroi, N. Sayo, T. Miura and H. Kumobayashi, Adv. Synth. Catal., 2001, 343, 264 CrossRef CAS.
- J.-P. Genet, T. Ayad and V. Ratovelomanana-Vidal, Chem. Rev., 2014, 114, 2824 CrossRef CAS PubMed.
- The cyclization of 2,2-diphenyl-5-hexenoic acid leading to the corresponding δ-lactone did not take place.
- The absolute configuration of 2h obtained with (R)-DTBM-segphos was determined to be R by comparison of the optical rotation with the reported one. J. Yang and N. Yoshikai, J. Am. Chem. Soc., 2014, 136, 16748 CrossRef CAS PubMed.
- F. Antonietti, E. Brenna, C. Fuganti, F. G. Gatti, T. Giovenzana, V. Grande and L. Malpezzi, Synthesis, 2005, 1148 CAS.
- R. H. Crabtree and G. E. Morris, J. Organomet. Chem., 1977, 135, 395 CrossRef CAS.
- T. Yamagata, H. Tadaoka, M. Nagata, T. Hirao, Y. Kataoka, V. Ratovelomanana-Vidal, J. P. Genet and K. Mashima, Organometallics, 2006, 25, 2505 CrossRef CAS.
- The stereochemistry of the major isomer was tentatively assigned by comparison of the chemical shifts of the hydride peaks with the reported values of [IrHCl(OAc)((S)-binap)]: see the ESI†.
- See the ESI† for details.
- K. Tsuchikama, M. Kasagawa and T. Shibata, Org. Lett., 2009, 11, 1821 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data. See DOI: 10.1039/c5cc05393e |
| This journal is © The Royal Society of Chemistry 2015 |