
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterfacial thiol–isocyanate reactions for functional nanocarriers: a facile route towards tunable morphologies and hydrophilic payload encapsulation†
Sören
Kuypers
ab,
Sumit Kumar
Pramanik
ab,
Lien
D'Olieslaeger
a,
Gunter
Reekmans
b,
Martijn
Peters
b,
Jan
D'Haen
ac,
Dirk
Vanderzande
bc,
Tanja
Junkers
bc,
Peter
Adriaensens
bc and
Anitha
Ethirajan
*ab
aInstitute for Materials Research (IMO), Hasselt University, Wetenschapspark 1, 3590 Diepenbeek, Belgium. E-mail: anitha.ethirajan@uhasselt.be
bOrganic and (Bio-)Polymer Chemistry, Institute for Materials Research, Hasselt University, Agoralaan D, 3590 Diepenbeek, Belgium
cIMEC, Associated lab IMOMEC, Wetenschapspark 1, 3590 Diepenbeek, Belgium
First published on 4th September 2015
Abstract
Functional nanocarriers were synthesized using an in situ inverse miniemulsion polymerization employing thiol–isocyanate reactions at the droplet interface to encapsulate hydrophilic payloads. The morphology of the nanocarriers is conveniently tunable by varying the reaction conditions and the dispersions are easily transferable to the aqueous phase.
One of the major challenges in developing nanocarriers is their design towards the ability to encapsulate hydrophilic substances. The encapsulation of hydrophilic compounds is highly valuable but at the same time also a very demanding task1 especially when the final purpose of the carriers is envisaged for an aqueous environment (e.g. the fields of drug delivery or bio-imaging to name a few).
Of the several heterophase polymerization techniques, miniemulsion has taken a fortified position as a versatile synthesis technique, allowing for effective encapsulation of both hydrophobic as well as hydrophilic compounds.2,3 It was previously shown that hydrophilic substances can be successfully encapsulated by using a nano-precipitation process or by interfacial reactions such as step growth, radical or anionic polymerization in inverse miniemulsion.4,5
The versatility of the thiol functionality to participate in different very efficient chemical reactions has seen tremendous focus recently for the generation of materials with interesting physical properties. In this regard, the thiol–ene click reaction6 has attracted significant interest owing to its high selectivity, efficiency and ability to use mild reaction conditions. Thiol–ene chemistry has – among other applications – been widely employed to obtain networks with tunable network properties.7,8 Lately, particles/capsules have also been reported based on this type of chemistry.9–11 Another thiol reaction that holds high potential, but has not been fully explored despite the fact that it is known since the late 1950s,12 is the thiol–isocyanate (thiol–NCO) conjugation; this reaction has to date remained largely unexploited especially for synthesizing nanomaterials. The nucleophilic addition of thiols to isocyanates yields thiourethane linkages. In the presence of a base catalyst, this equimolar reaction is facilitated through the generation of a strong nucleophilic thiolate ion and an electron deficient carbonyl carbon at the isocyanate. This reaction proceeds on a fast time scale (faster than classical urethane formation), at ambient temperature and affords high conversions. The reaction mechanism is given in Fig. 1. The thiourethane (–NH–CO–S–) functionality is a sulphur analogue of urethane (–NH–CO–O–) and the former's incorporation into the polymer chain is known to introduce interesting mechanical and thermal properties.13,14 Also, the high refractive index values of polythiourethanes make the latter highly appealing for optical and coating applications as compared to their polyurethane counterparts.15,16 Shin et al. made ternary networks using thiol–isocyanate–ene sequential/simultaneous click reactions where an amine catalyst was used to trigger the thiol–isocyanate reaction and ultraviolet (UV) light was used to trigger a radical thiol–ene reaction, resulting in a thiourethane/thiol–ene hybrid network.17 Matsushima et al. also used thiol-click chemistry to create thiol–isocyanate–acrylate ternary networks.18 Previously, thiol–NCO chemistry was used in a modular approach for a rapid and robust fabrication of highly functional, multicomponent polymer brush surfaces19 and lately for developing self-healing epoxy thermosets.20 Recently, microparticles (40–250 μm) were synthesized using nucleophile-catalyzed thiol–NCO reaction in water employing microfluidics.21 With the recent revival of the thiol–NCO reaction towards interesting applications, the potential of this reaction towards designing nanomaterials with functional properties is highly desirable. Though polyaddition reactions between diisocynates and diols/diamines have been extensively studied for nanocarrier formation, to the best of our knowledge, isocyanate–thiol interfacial reactions have not been reported to date. The latter reaction offers materials with different useful properties as compared to classical urethanes and will hence lead to novel functional nanocarriers.
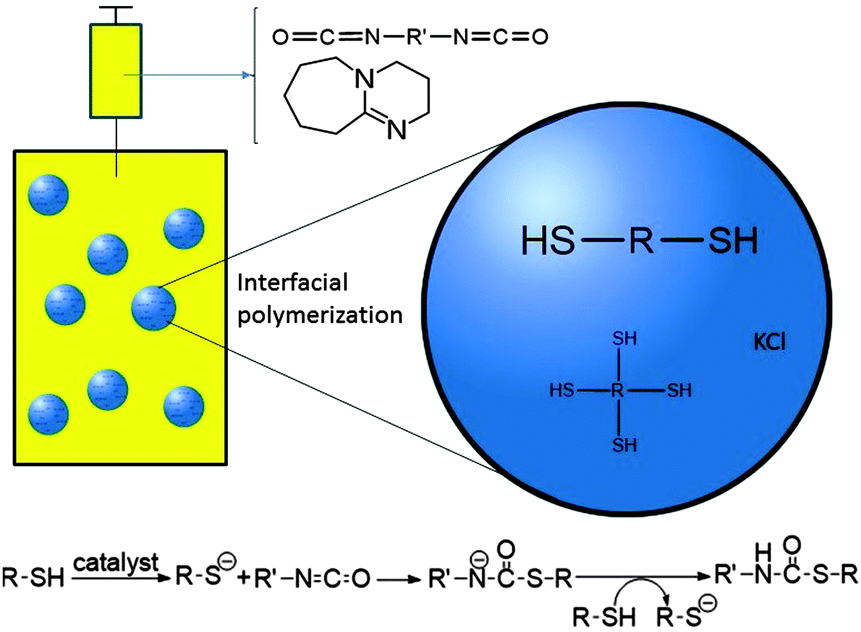 | ||
| Fig. 1 Schematic representation of a base catalyzed interfacial polymerization reaction between diisocyanate and (tetra-)dithiol monomers in an inverse miniemulsion. | ||
In this work, for the first time the miniemulsion technique has been used for in situ thiol–NCO reactions at the droplet interface for the design of nanocarriers containing thiourethane linkages (Fig. 1). For encapsulation of a hydrophilic payload using inverse miniemulsion, a 1 M KCl solution was used as a model substance. Nanocarriers were formulated at room temperature using 1,8-diazabicycloundec-7-ene (DBU) as a base catalyst and employing different stoichiometric ratios of 1,4-butanedithiol (BDT) and toluene diisocyanate (TDI) as the respective bi-functional monomers. In order to impart potential post-surface grafting possibilities to the nanocarriers, pentaerythritol tetra-3-mercaptopropionate (PETMP), a multifunctional crosslinker monomer, was also used in the reaction formulations. The detailed synthesis procedure is presented in the ESI.† Briefly, cyclohexane was used as the continuous phase and the dispersed phase employed consists of thiol monomer(s), DMSO, and KCl solution. After pre-emulsification followed by sonication, stable nanodroplets of the dispersed phase were obtained. Subsequently, an additive phase containing the catalyst and the TDI monomer in cyclohexane was slowly introduced into the continuous phase and then the reaction mixture was left for stirring.
As the reaction conditions have a significant effect on the nanocarrier morphology, the influence of the different reaction parameters – the amounts of monomers, the presence of a crosslinker and a catalyst – on the resulting morphology was studied systematically. Since, the reaction in the absence of a catalyst at room temperature is not feasible, the same reactions were also repeated using elevated temperature (60 °C) in order to study the effect of the catalyst on the morphology. For all formulations used, the stability of the resulting dispersions was first checked optically for any visible phase separation and samples were then directly used for further characterization. The colloidal stability, the size and polydispersity index (PDI) of the samples were studied using dynamic light scattering (DLS). The reaction efficiency was studied thermogravimetically in the form of solid content of the obtained dispersion. Chemical analyses of the insoluble products were performed using high resolution solid state 13C nuclear magnetic resonance (NMR) and Fourier-transform infrared (FT-IR) spectroscopy. As a proof-of-concept for the use of such nanocarriers in biomedical applications, biocompatibility studies were also performed. The dispersion characteristics of the different samples tested are presented in Table 1.
| Samplea | Dispersed phase monomer(s) | Additive phase | Size (nm)/PDI | Expt. solid content (%) | Theo. solid content (%) |
|---|---|---|---|---|---|
| a Samples 1–4 were prepared using a catalyst at room temperature, while, samples 5–8 were obtained at 60 °C (for details see the ESI). | |||||
| 1 | 2 mmol BDT | 2 mmol TDI, DBU | 173/0.04 | 5.4 | 7.1 |
| 2 | 4 mmol BDT | 4 mmol TDI, DBU | 193/0.12 | 7.4 | 11.9 |
| 3 | 4 mmol BDT, 0.1 mmol PETMP | 4.2 mmol TDI, DBU | 200/0.14 | 11.8 | 13.7 |
| 4 | 4 mmol BDT, 0.2 mmol PETMP | 4.4 mmol TDI, DBU | 182/0.26 | 11.9 | 14.3 |
| 5 | 2 mmol BDT | 2 mmol TDI | 168/0.09 | 5.3 | 7.2 |
| 6 | 4 mmol BDT | 4 mmol TDI | 188/0.12 | 8.0 | 12.9 |
| 7 | 4 mmol BDT, 0.1 mmol PETMP | 4.2 mmol TDI | 221/0.19 | 13.1 | 13.6 |
| 8 | 4 mmol BDT, 0.2 mmol PETMP | 4.4 mmol TDI | 202/0.24 | 12.8 | 14.3 |
From the hydrodynamic diameters (intensity average), it can be seen that most of the synthesized nanocarriers have a size range between 165 and 225 nm. From the obtained values, a trend between the size and the amount of monomer used in the reaction can be observed; the size of the nanocarriers mostly increases with the increase in the monomer amount. It can also be seen that the polydispersity index (PDI) is higher when crosslinkers were employed for samples prepared using both a base catalyst as well as elevated temperature. The increased size and the PDI observed for the reactions performed in the presence of the crosslinker could be attributed to the structure of the tetrafunctional crosslinker, which is more bulky than the linear bi-functional monomer. The sizes observed here in general are comparable and similar to values reported for polyurethane capsules.22
The experimental solid content here refers to the dispersion solid content containing only particulates, excluding any large aggregates/bulk materials formed during reaction. The theoretical solid content refers to the calculated dispersion solid content upon full conversion of all monomers added to the reaction. The solid contents measured are also in accordance with literature values reported for polyurethane capsules.22 The solid contents are higher when the monomer amounts are increased. This trend is as expected as higher amounts of starting reactants lead to an increased product formation. The overall success of the thiol–NCO reaction is determined by the presence of the thiourethane groups in the product. In Fig. 2, the NMR data obtained for selected samples are depicted. Aliphatic carbons are located in the range between 15 and 45 ppm. The methyl group originating from the diisocyanate is assigned to the peak at 18 ppm. The shoulders at 26 ppm and 64 ppm originate for the crosslinker. The peak at 70 ppm represents the alcohol and the ether functionalities of the surfactant; the latter is a block copolymer consisting of polyhydroxystearic acid and polyethylene glycol moieties.23,24 The peaks ranging from 140 ppm to 110 ppm originate from the aromatic ring of the diisocyanate used for polymerization. The small peak at 154 ppm corresponds to urea–urethane.25 The formation of urea can be attributed to the reaction of isocyanate with amine groups formed in the first place by the hydrolysis of the highly reactive isocyanate with ambient water. Such an observation has been reported in the case of formation of polyurethane nanocapsules before.26 Urethane formation is also possible due to the alcohol functionality present in the surfactant (see Fig. S6 in the ESI†). Which of the two reactions is predominating as side reaction is purely based on NMR is difficult to verify. Regardless, a well-defined intense peak at 166 ppm confirms the successful formation of thiourethane.27 FT-IR analysis might aid in exact structure elucidation and spectra for the same selected samples as in Fig. 2 are presented in Fig. 3. The full spectral range for all samples in Table 1 (Fig. S7, ESI†), monomers (Fig. S2–S4, ESI†) and products from an analogue bulk reaction (Fig. S5, ESI†) are presented in the ESI.†
 | ||
| Fig. 2 High resolution solid state 13C NMR data showing the presence of thiourethane for samples 4 (a), 8 (b), 2 (c) and 6 (d) (see Table 1). | ||
 | ||
| Fig. 3 Transmission FT-IR spectra showing the presence of thiourethanes for samples 4 (a), sample 8 (b), sample 2 (c) and sample 6 (d). | ||
The disappearance of the functional groups stemming from the monomers is depicted in Fig. S7 in the ESI† by the disappearance of the characteristic isocyanate and thiol peaks (2280 cm−1 to 2270 cm−1 and 2556 cm−1 to 2400 cm−1, respectively).19 The presence of the thiourethane peak at ∼1680 cm −1 in Fig. 3 indicates the successful reaction between the diisocyanate and the thiol moieties.27,28 It can be seen that the thiourethane peaks are broadened due to the presence of urea at ∼1640 cm−1.26,29 The urea is formed from the reaction between isocyanate end groups and water as indicated above. Since the reaction is performed under equimolar conditions and the signal of the diisocyanate monomers is no longer present in the FT-IR data, it can be assumed that the excess thiol groups present in the reaction medium have been oxidized to form disulphides.30 Consequently, the presence of urethane at ∼1700 cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26,28,29 could not be ascertained by the FT-IR data. This is likely due to only trace amounts of urethane being formed owing to the small amount of surfactants used (see Fig. S6 in the ESI†). In addition, as the amines are more reactive, the consumption of isocyanates for urea formation might also affect the urethane formation.27 Therefore, the signal at 154 ppm in the NMR spectrum is in all likelihood mainly due to the formation of urea rather than urethane moieties. The NMR results together with the FT-IR data thus clarify the presence of thiourethane–urea linkages in the samples.
26,28,29 could not be ascertained by the FT-IR data. This is likely due to only trace amounts of urethane being formed owing to the small amount of surfactants used (see Fig. S6 in the ESI†). In addition, as the amines are more reactive, the consumption of isocyanates for urea formation might also affect the urethane formation.27 Therefore, the signal at 154 ppm in the NMR spectrum is in all likelihood mainly due to the formation of urea rather than urethane moieties. The NMR results together with the FT-IR data thus clarify the presence of thiourethane–urea linkages in the samples.
The nanocarrier morphology was studied using transmission electron microscopy (TEM). The size and size distribution (qualitatively) observed in the TEM images (Fig. 4) for all samples are in agreement with the size and polydispersity measured using DLS. As it can be seen in Fig. 4a in the case of base catalysed reactions, sample 1 (without a crosslinker) gave a homogeneous bulk nanoparticle morphology. While sample 4 (Fig. 4b) (with a crosslinker) clearly afforded a core–shell nanocapsule morphology (for sample 3 see Fig. S8 in the ESI†). The reaction using elevated temperatures conducted in the absence of the catalyst and without a crosslinker (sample 6) yielded nanoparticles (Fig. 4c). In stark contrast to samples 3 and 4, in Fig. 4d (sample 8) the addition of the crosslinker to the elevated temperature reaction also resulted in nanoparticles (for sample 7 see Fig. S9 in the ESI†). In the case of nanocapsules the average shell thicknesses are 37.2 ± 7 nm and 51 ± 8 nm for samples 3 and 4 respectively. With increasing amount of cross-linkers the shell-thickness increases and also the size distribution (see Fig. S10 in the ESI†) becomes relatively broader reflecting the trend in DLS results. The successful encapsulation of KCl solution was confirmed by the presence of dark spots (salt crystals) within the capsules in the TEM images and was also confirmed by energy-dispersive X-ray spectroscopy (EDX) (see Fig. S11 in the ESI†).
 | ||
| Fig. 4 TEM images of samples 1 (a), 4 (b), 6 (c) and 8 (d) from the organic phase (scale bar 500 nm). Images (a) and (b) are of samples employing base catalyst, while images (c) and (d) are of samples prepared at elevated temperature. Images e and f (scale bar 200 nm) are of samples 3 and 4, respectively, after redispersion in water. | ||
To test the potential of these nanocarriers for biomedical applications, the synthesized nanocapsules have been transferred from the oil phase to a water phase. The redispersed aqueous dispersions were colloidally stable and the hydrodynamic diameters measured using DLS were 267 nm with a PDI value of 0.186 and 297 nm with a PDI value of 0.177 for samples 3 and 4 respectively. The significant increase in size after redispersion can be attributed to the pronounced hydration of the hydrophilic part of the block copolymeric surfactant in the aqueous phase. Using the TEM studies (Fig. 4e and f), the capsule morphology for samples with different crosslinker amounts can be clearly seen where the shell remains intact and evidently unaffected by the redispersion process. Using the EDX studies (see Fig. S12 in the ESI†), the presence of salt crystals (seen as dark spots within the capsules) was ascertained. Additionally, there was no sign of large aggregates of the nanocarriers (broken capsule debris) in the overview inspection of the TEM grid. Thus, DLS measurements and TEM characterization unambiguously confirm that aqueous dispersion of nanocapsules containing hydrophilic payload can be achieved successfully. Additionally, biocompatibility studies using Alamar blue assay performed on the redispersed samples (see Fig. S13 in the ESI†) clearly indicate that the nanocarriers are fully biocompatible thereby making them excellent candidates for biomedical applications. As a proof-of-concept, doxorubicin, a potent anticancer drug was encapsulated (see Fig. S14 in the ESI†).
For the first time thiourethane-based nanocarriers encapsulating hydrophilic substances have been synthesized via the inverse miniemulsion technique using an in situ thiol–NCO reaction at the droplet interface. The presence of thiourethane functionality was confirmed by FT-IR spectroscopy and high resolution 13C solid-state NMR spectroscopy. Also, side reactions leading to urea formation were ascertained by FT-IR. The morphology of the nanocarriers could be conveniently tuned by adjusting the reaction conditions as confirmed by TEM imaging. The presence of catalyst allows for the formation of tunable morphologies depending on the choice of the monomers used (i.e. with or without a crosslinker). The tunability of morphology offers designing nanocarriers for the desired purpose. For instance, nanocapsule morphology (with crosslinker) is suited for applications where a large aqueous core to polymer ratio encapsulating a large payload is required and the bulk particle morphology (without crosslinker) for applications envisaging high polymer content. The nanocapsules were also conveniently transferred to an aqueous phase while keeping their shell intact and were also successfully tested for their biocompatibility. With a suitable choice of the monomers, surface functionalization using the versatile thiol and isocyanate groups will be feasible. The shell is currently being modified to impart biodegradability by varying the monomer choices and to functionalize using post-modification steps (via the tetrafunctional thiol moiety). The shells will be further studied for their physical properties and any possible leakage. In concise, we could successfully demonstrate that thiol–NCO reaction at the interface opens new possibilities for designing functional nanocarriers for biomedical applications.
The authors thank BOF funding of Hasselt University and the support from BELSPO via IAP Programme (P7/05). The authors are thankful to Prof. M. Van Bael for the access to the DLS device. A.E. is a FWO (Fonds Wetenschappelijk Onderzoek) postdoctoral fellow. M.P. is funded by “Agency for Innovation by Science and Technology” in Flanders (IWT).
Notes and references
- S. Vrignaud, J. P. Benoit and P. Saulnier, Biomaterials, 2011, 32, 8593–8604 CrossRef CAS PubMed.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- A. Ethirajan and K. Landfester, Chem. – Eur. J., 2010, 16, 9398–9412 CrossRef CAS PubMed.
- M. Antonietti and K. Landfester, Prog. Polym. Sci., 2002, 27, 689–757 CrossRef CAS.
- J. M. Siebert, G. Baier, A. Musyanovych and K. Landfester, Chem. Commun., 2012, 48, 5470–5472 RSC.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- N. Zaquen, B. Wenn, K. Ranieri, J. Vandenbergh and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 178–187 CrossRef CAS PubMed.
- J. Vandenbergh, K. Ranieri and T. Junkers, Macromol. Chem. Phys., 2012, 213, 2611–2617 CrossRef CAS PubMed.
- D. V. Amato, D. N. Amato, A. S. Flynt and D. L. Patton, Polym. Chem., 2015, 6, 5625–5632 RSC.
- C. Fleischmann, J. Gopez, P. Lundberg, H. Ritter, K. L. Killops, C. J. Hawker and D. Klinger, Polym. Chem., 2015, 6, 2029–2037 RSC.
- U. Paiphansiri, G. Baier, A. Kreyes, D. Yiamsawas, K. Koynov, A. Musyanovych and K. Landfester, Macromol. Chem. Phys., 2014, 215, 2457–2462 CrossRef CAS PubMed.
- R. G. Arnold, J. A. Nelson and J. J. Verbanc, Chem. Rev., 1957, 57, 47–76 CrossRef CAS.
- E. Dyer and D. W. Osborne, J. Polym. Sci., 1960, 47, 361–371 CrossRef CAS PubMed.
- O. D. McNair, D. P. Brent, B. J. Sparks, D. L. Patton and D. A. Savin, ACS Appl. Mater. Interfaces, 2014, 6, 6088–6097 CAS.
- A. Kultys, M. Rogulska and S. Pikus, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1770–1782 CrossRef CAS PubMed.
- S. S. Naik, J. W. Chan, C. Comer, C. E. Hoyle and D. A. Savin, Polym. Chem., 2011, 2, 303–305 RSC.
- J. Shin, H. Matsushima, C. M. Comer, C. N. Bowman and C. E. Hoyle, Chem. Mater., 2010, 22, 2616–2625 CrossRef CAS.
- H. Matsushima, J. Shin, C. N. Bowman and C. E. Hoyle, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3255–3264 CrossRef CAS PubMed.
- R. M. Hensarling, S. B. Rahane, A. P. LeBlanc, B. J. Sparks, E. M. White, J. Locklin and D. L. Patton, Polym. Chem., 2011, 2, 88–90 RSC.
- X. K. D. Hillewaere, R. F. A. Teixeira, L.-T. T. Nguyen, J. A. Ramos, H. Rahier and F. E. Du Prez, Adv. Funct. Mater., 2014, 24, 5575–5583 CrossRef CAS PubMed.
- J. Tan, C. Li, H. Li, H. Zhang, J. Gu, B. Zhang, H. Zhang and Q. Zhang, Polym. Chem., 2015, 6, 4366–4373 RSC.
- E.-M. Rosenbauer, M. Wagner, A. Musyanovych and K. Landfester, Macromolecules, 2010, 43, 5083–5093 CrossRef CAS.
- N. Yasarawan and J. S. van Duijneveldt, Soft Matter, 2010, 6, 353–362 RSC.
- E. Lovelady, S. D. Kimmins, J. Wu and N. R. Cameron, Polym. Chem., 2011, 2, 559–562 RSC.
- S. M. Clift, J. Grimminger, K. Muha, presented in part at the SPI Conference, 1994.
- U. Paiphansiri, J. Dausend, A. Musyanovych, V. Mailänder and K. Landfester, Macromol. Biosci., 2009, 9, 575–584 CrossRef CAS PubMed.
- J. D. Flores, J. Shin, C. E. Hoyle and C. L. McCormick, Polym. Chem., 2010, 1, 213–220 RSC.
- J. F. Smith and E. C. Friedrich, J. Am. Chem. Soc., 1959, 81, 161–163 CrossRef CAS.
- F. Tiarks, K. Landfester and M. Antonietti, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2520–2524 CrossRef CAS PubMed.
- V. Dritsa, K. Pissaridi, E. Koutoulakis, I. Mamarelis, C. Kotoulas and J. Anastassopoulou, In Vivo, 2014, 28, 91–98 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, synthesis procedure, characterization methods, sample details, FT-IR spectra, TEM images and EDX spectra of particles, and biocompatibility studies. See DOI: 10.1039/c5cc05258k |
| This journal is © The Royal Society of Chemistry 2015 |