
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile access to a heterocyclic, sp3-rich chemical scaffold via a tandem condensation/intramolecular nitrone–alkene [3+2] cycloaddition strategy†
M. J.
Rawling
a,
T. E.
Storr
a,
W. A.
Bawazir
ab,
S. J.
Cully
a,
W.
Lewis
a,
M. S. I. T.
Makki
b,
I. R.
Strutt
c,
G.
Jones
c,
D.
Hamza
c and
R. A.
Stockman
*a
aSchool of Chemistry, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: robert.stockman@nottingham.ac.uk; Fax: +44 (0)115 951356
bDepartment of Chemistry, Faculty of Science, King Abdul-Aziz University, P.O. Box 80203, Jeddah 21315, Saudi Arabia
cSygnature Discovery Limited, BioCity, Pennyfoot Street, Nottingham, NG7 1GF, UK
First published on 9th July 2015
Abstract
A heterocyclic, sp3-rich chemical scaffold was synthesised in just 6 steps via a highly regio- and diastereo-selective tandem nitrone formation/intramolecular nitrone–alkene [3+2] cycloaddition reaction. A library of 543 lead-like compounds based on the scaffold core has been produced.
Libraries of small molecules capable of exploring novel, three-dimensional chemical space are highly desired in early-stage probe- and drug-discovery screening programmes.1 Multiple authors have suggested that molecules containing a high fraction of sp3-hybridised carbon atoms (Fsp3) and numerous stereocentres have a lower rate of attrition during each stage of the drug discovery and development process once a hit has been established, thus potentially providing higher-quality lead compounds.2,3 However, the development of facile synthetic routes to libraries of stereochemically-rich molecules with a high Fsp3 remains a formidable challenge for the chemical community.4
Over the past decade our group has been interested in the rapid synthesis of architecturally complex molecules from simple, symmetrical starting materials, through de-symmetrising tandem reactions.5 This has led to the efficient synthesis of a number of natural products including xenovenine (4 steps),6 alkaloid cis-223B (3 steps),6,7 histrionicotoxin (9 steps),8 anatoxin-a (10 steps),9 hippodamine (8 steps) and epi-hippodamine (11 steps).10 We have recently demonstrated that through tandem reactions involving key complexity-generating, ring-forming operations, such as [3+2] dipolar cycloadditions, densely-functionalised tricyclic cores containing a spirocyclic centre can be generated in a diastereoselective manner.5,11,12
A current focus within our research group is the exploitation of the knowledge gained throughout these investigations, in the efficient synthesis of sp3-rich chemical scaffolds for drug discovery.13 Polycyclic scaffolds containing a central spiro-ring fusion are desirable in drug discovery due to their structural rigidity resulting in a reduced conformational entropy penalty upon ligand–protein binding.14 In addition, incorporation of a spiro-centre often provides greater structural novelty and enhanced physical properties over that of flat, (hetero)aromatic compounds.2,3 To maximise its potential a quality chemical scaffold should: (i) contain multiple points of diversity, (ii) be ‘lead-like’,15 with a sufficiently low molecular weight (≤300 Da) and cLog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (≤3) to allow derivatisation of the scaffold core into a library of compounds that largely adheres to Lipinski's Rule of Five,16 and (iii) not contain any metabolically labile moieties or toxicophores. From a practical standpoint, the synthetic route to a scaffold should be concise, reliable, safe, and scalable to provide rapid access to large quantities of material.
P (≤3) to allow derivatisation of the scaffold core into a library of compounds that largely adheres to Lipinski's Rule of Five,16 and (iii) not contain any metabolically labile moieties or toxicophores. From a practical standpoint, the synthetic route to a scaffold should be concise, reliable, safe, and scalable to provide rapid access to large quantities of material.
Following these guiding principles, we report the facile and diastereoselective synthesis of a novel, heterocyclic chemical scaffold 1 (Fig. 1). The rigid, tricyclic core of scaffold 1 contains four contiguous stereocentres with one spirocyclic centre, and has a high degree of bond saturation,2 with an Fsp3 value of 0.9. The scaffold 1 has three points of diversity (R1, R2 and R3) to allow exploration of chemical space along three distinct vectors. The low molecular weight (196 Da) and cLogP (−1.2) of the unsubstituted scaffold 1 (R1, R2, R3 = H) provides an excellent platform for diversification into a library of drug-like molecules, for screening against various biological targets.
 | ||
| Fig. 1 Retrosynthetic analysis of scaffold 1. | ||
On the basis of previous work, we envisioned that the 3-hydroxy lactam unit of scaffold 1 could be derived from key intermediate isoxazolidine 2, by N–O bond reduction followed by cyclisation (Fig. 1).5,12 It was proposed that spirocyclic isoxazolidine 2 could be obtained diastereoselectively through a tandem nitrone formation/intramolecular nitrone–alkene [3+2] cycloaddition from piperidinone 3, in a single complexity-generating step. Mono-alkylated piperidinone 3 should be readily accessible from a suitable N-protected piperidinone 4.
N-Benzyl piperidinone 4a was selected as the starting point in the synthesis of scaffold 1 due to its commercial availability and low cost. Piperidinone 4a was alkylated via hydrazone 5a to avoid the well-documented issues associated with the direct alkylation of ketones (Scheme 1).17 Treatment of piperidinone 4a with a small excess of N,N-dimethylhydrazine (1.2 equiv.) under Dean–Stark conditions gave hydrazone 5a in quantitative yield after 2 hours, with no need for purification.18 α-Deprotonation of hydrazone 5a with nBuLi (1.1 equiv.) then C-alkylation with 4-bromo-1-butene (1.5 equiv.) and in situ hydrolysis of the hydrazone moiety (2 M HCl), gave previously unreported homoallylated piperidinone 6 in excellent overall yield (81%). This one-pot procedure was typically performed on large scale, yielding up to 24 g of piperidinone 6 in a single batch. Unexpectedly, attempted alkylation of the carbamate-protected piperidinone analogues 4b (N-Boc) and 4c (N-Cbz) resulted in recovered starting material. Further investigation revealed that hydrazones 5b and 5c had formed without incident. However, α-deprotonation of hydrazones 5b and 5c using either LDA or nBuLi had not occurred, as quenching the reaction with D2O revealed no deuterium incorporation by mass spectrometry or 1H NMR spectroscopy. Common additives (LiCl, TMEDA and DMPU) for use with lithium bases were employed,19 but failed to have any effect. The reason for the apparent lack of reactivity of N,N-dimethylhydrazones 5b and 5c to deprotonation is currently unknown, but was not explored further.20
 | ||
| Scheme 1 Synthesis of key intermediate isoxazolidine 2. | ||
The trans-α,β-unsaturated ester moiety was incorporated into N-benzyl piperidinone 3a by cross-metathesis between ethyl acrylate 7 and the terminal olefin of 6. Typical Grubbs cross-metathesis conditions (Grubbs II [10 mol%], ethyl acrylate 7 [8 equiv.], CH2Cl2, 20 °C)21 gave α,β-unsaturated ester 3a in a disappointing 32% yield (E:Z 9:1) after 4 days, which could not be improved by varying the catalyst, reagent stoichiometry, solvent or temperature. In an effort to improve both the yield and reaction rate, the ‘Copper Iodide Effect’ was utilised, as described by Lipshutz and co-workers.22 Addition of copper iodide (3 mol%) as co-catalyst gave α,β-unsaturated ester 3a (E:Z 11:1) in 89% isolated yield in only 3 hours on large scale (>10 g), representing a dramatic increase in conversion and reaction rate. Beneficially, the procedure also required considerably less Grubbs II catalyst (1.5 mol%) and ethyl acrylate 7 (3 equiv.) and was performed in ethereal solvent, rather than chlorinated media, deeming it more attractive from a financial, safety and environmental perspective.23 The two geometric isomers of α,β-unsaturated ester 3a were separated by silica gel column chromatography and the major (E)-isomer taken forward in the synthesis.
Treatment of piperidinone 3a with N-benzylhydroxylamine hydrochloride (1.5 equiv.) in the presence of NaOAc (3.0 equiv.) in refluxing toluene afforded key intermediate tricyclic isoxazolidine 2a in 66% isolated yield (Scheme 1). In a single complexity-generating operation, three new stereogenic centres and two new rings were formed, to generate a fused, tricyclic, spiro-isoxazolidine. Subjecting N-methylhydroxylamine hydrochloride to the same reaction conditions resulted in the analogous isoxazolidine 2b in 61% isolated yield, showing that one point of diversity can be incorporated into the scaffold at this stage. Significantly, only a single regio- and diastereo-isomer was observed by 1H NMR spectroscopy in both cases.
The tandem reaction of piperidinone 3a to isoxazolidine 2b initially proceeds through condensation of 3a with the mono N-substituted hydroxylamine to give nitrone 8 (Fig. 2). Subsequent intramolecular nitrone–alkene [3+2] cycloaddition gives two possible regioisomeric isoxazolidine products, 2b or 9. Computational analysis of isoxazolidines 2b and 9 (Hartree–Fock 6-31G*) revealed a small energy difference between the two isomers, with the unobserved 6,6,5 ring system 9 being 4.62 kJ mol−1 lower in energy than the observed 6,5,5 ring system 2b. However, the transition state 8-TS2 during formation of the lower energy 6,6,5 ring system 9 would experience substantial unfavourable steric interactions arising from the close proximity of the ester function to the developing congested spirocentre. Conversely, the transition state 8-TS1 in the formation of the 6,5,5 ring system 2b has the ester moiety located away from the developing spirocentre, resulting in minimal steric interactions and therefore a more favourable reaction pathway. Attempted isomerisation of 2b to the energetically favoured isoxazolidine 9 (μW, PhCl, 200 °C, 2 hours) resulted in a complex mixture of starting material 2b and decomposition products.
 | ||
| Fig. 2 Formation of regioisomeric isoxazolidines 2b and 9. | ||
Treatment of isoxazolidines 2a and 2b with activated zinc in aqueous acetic acid prompted smooth transformation to the corresponding 3-hydroxy lactams 11a and 11b in excellent yield (93% and 88% respectively) with no need for purification (Scheme 2). Mechanistically, reduction of the N–O bond in isoxazolidine 2, followed by C–C bond rotation to 10, allowed ring-closure to lactam 11.
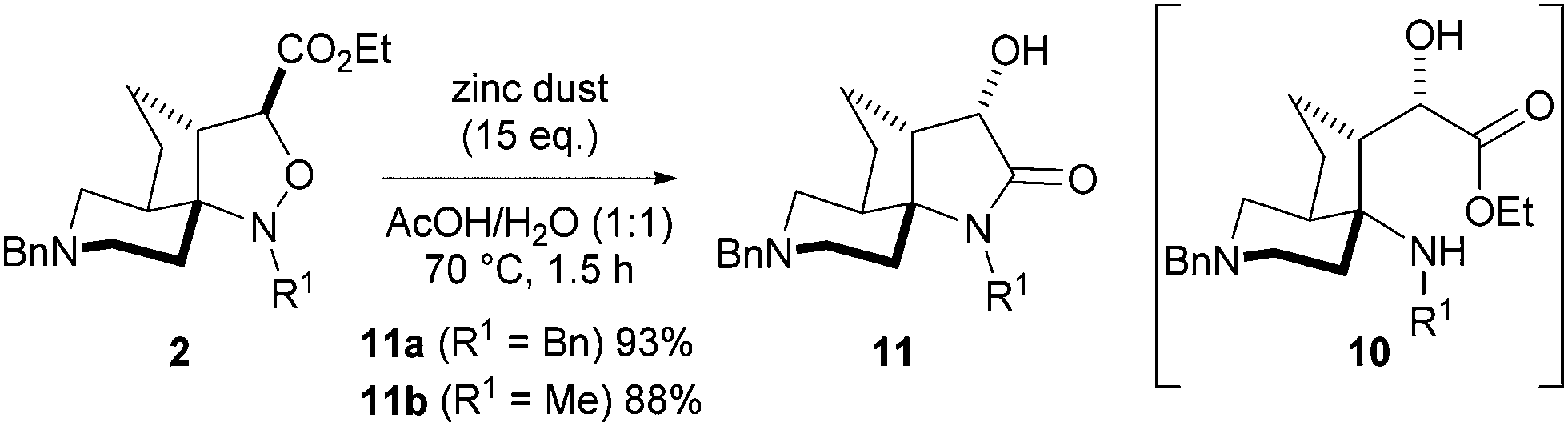 | ||
| Scheme 2 Reductive cyclisation of isoxazolidine 2 to 3-hydroxy lactam 11. | ||
X-Ray crystallographic analysis of lactams 11a and 11b confirmed the atom connectivity and relative stereochemistry (Fig. 3). The X-ray images clearly show that the three points of diversification on scaffolds 11a (atoms N1, N7 and O21, Fig. 3) and 11b (atoms N1, N7 and O22, Fig. 3) will allow library synthesis in three defined and divergent trajectories, providing substantial exploration of three-dimensional chemical space.
 | ||
| Fig. 3 X-ray crystallographic images of 3-hydroxy lactams 11a and 11b. | ||
With the scaffold core structure 11 in hand, it was important to demonstrate that the remaining two points of diversity on 1 (R2 and R3, Fig. 1) were capable of being functionalised without complication, prior to full library synthesis being undertaken. O-Alkylation of 11 was exemplified by deprotonation of the hydroxy function with sodium hydride, then treatment with stoichiometric iodomethane to give methyl ethers 12a and 12b in 65% and 61% isolated yield respectively (Scheme 3).
 | ||
| Scheme 3 O-Alkylation and N-debenzylation. | ||
Deprotection of the piperidyl N-benzyl moiety of scaffolds 11a, 11b, 12a and 12b occurred cleanly using Pearlman's catalyst under an atmospheric pressure of hydrogen gas to give the secondary amines 13a–13d in excellent yield (88–93%, Scheme 3). Interestingly, the lactam N-benzyl group of 11a and 12a was not cleaved under these conditions, presumably due to steric hindrance around the N–Bn bond imposed by the adjacent spirocentre. Remarkably, increasing the hydrogen pressure (50 atm, 4 days), addition of acid (AcOH or HCl) or employing typical transfer hydrogenation conditions (Pd/C, ammonium formate, MeOH, reflux, 24 h) also proved ineffective at lactam N-debenzylation in 11a (R1, Scheme 2).
The deprotected piperidyl nitrogen atom (N1, 11, Fig. 3) represented the main library diversification point on scaffold 1. Secondary amines can undergo an array of useful transformations for library synthesis, including (i) coupling with carboxylic acids to make amides,24 (ii) reaction with isocyanates to form ureas, (iii) reductive amination with aldehydes or ketones to give tertiary amines,25 and (iv) treatment with sulfonyl chlorides to make sulfonamides. A small, exemplary library of compounds was produced in good yield (30–72%) using typical reductive amination (conditions A), sulfonylation (conditions B) and amidation (conditions C) protocols (11b, 1a–e, Fig. 4), proving that the final functional handle of scaffold 1 (R3, Fig. 1) could be subjected to a variety of standard reactions without issue.
 | ||
| Fig. 4 Conditions A (reductive amination) – 13 (1.0 equiv.), ArCHO (1.5 equiv.), NaBH(OAc)3 (1.2 equiv.), AcOH (2.5 equiv.), 1,2-DCE or NMP, 18 °C, 18 h; conditions B (sulfonylation) – 13 (1.0 equiv.), ArSO2Cl (1.3 equiv.), pyridine, 18 °C, 18 h; conditions C (amidation) – 13 (1.5 equiv.), ArCO2H (1.0 equiv.), HATU (2.0 equiv.), Hünig's base (3.0 equiv.), DMF, 18 °C, 18 h. | ||
The synthetic route was scaled up to provide a total of 130 mmol of the scaffold core 11. Derivatisation of R1, R2 and R3 produced a library of 543 novel compounds (∼0.1 mmol each) to be incorporated into the European Lead Factory's screening programme. Computational assessment of the library indicates that it is largely Lipinski's Rule of Five compliant, with an average molecular weight of 429 and cLogP of 1.6 (Fig. 5). The library also has a high degree of three-dimensionality and bond-saturation, with an average Fsp3 of 0.54 (Fig. 5).
 | ||
| Fig. 5 Plot of cLogP versus MW (left) and Fsp3 analysis (right) of the 543-compound library based on scaffold 1. | ||
The development of efficient synthetic routes to libraries of small, sp3-rich molecules for use in drug discovery remains a challenging but important goal. We have reported the facile synthesis and diversification of a novel, heterocyclic chemical scaffold 1. The spirocyclic scaffold core 13 (R1 = Bn or Me, R2 = R3 = H) was accessed in only six operationally simple synthetic steps from commercially available N-benzyl piperidinone 4a, in an overall yield of 41% (13a) or 36% (13b) (averaging 87% or 85% yield per step, respectively). The synthetic route to scaffold 13 required just one protecting group manipulation (N-debenzylation), and can be completed in less than five working days on a multi-gram scale. A highly regio- and diastereo-selective tandem condensation/intramolecular nitrone–alkene [3+2] cycloaddition reaction provided key intermediate isoxazolidine 2 in a single, complexity-generating operation, from α-functionalised piperidinone 3a. Subsequent one-pot isoxazolidine N–O bond reduction and cyclisation gave the scaffold core structure 11, which was confirmed by X-ray crystallographic analysis. Diversification of the scaffold core has provided a library of 543 drug-like molecules, which will be screened against various biological targets as part of the European Lead Factory. Work is ongoing within our laboratory to develop efficient routes to novel sp3-rich chemical scaffolds for drug discovery.
This research was done within the European Lead Factory and has received support from the Innovative Medicines Initiative Joint Undertaking (grant no. 115489), with financial contribution from the European Union's Seventh Framework Programme (FP7/2007–2013) and EFPIA companies' in-kind contribution. The authors would like to thank Professor Christopher Moody and Tom McInally (University of Nottingham), in addition to Dr Iain Miller and Dr Geraint Jones (Sygnature Discovery Limited) for useful insight and discussion.
Notes and references
- J.-L. Reymond and M. Awale, ACS Chem. Neurosci., 2012, 3, 649 CrossRef CAS PubMed; A. W. Hung, A. Ramek, Y. Wang, T. Kaya, J. A. Wilson, P. A. Clemons and D. W. Young, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6799 CrossRef PubMed; J.-L. Reymond, R. van Deursen, L. C. Blum and L. Ruddigkeit, MedChemComm, 2010, 1, 30 RSC.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed.
- M. Aldeghi, S. Malhotra, D. L. Selwood and A. W. E. Chan, Chem. Biol. Drug Des., 2014, 83, 450 Search PubMed; F. Lovering, MedChemComm, 2013, 4, 515 RSC; P. A. Clemons, J. A. Wilson, V. Dancik, S. Muller, H. A. Carrinski, B. K. Wagner, A. N. Koehler and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6817 CrossRef CAS PubMed; T. J. Ritchie and S. J. F. MacDonald, Drug Discovery Today, 2009, 14, 1011 CrossRef PubMed; Y.-k. Kim, M. A. Arai, T. Arai, J. O. Lamenzo, E. F. Dean, III, N. Patterson, P. A. Clemons and S. L. Schreiber, J. Am. Chem. Soc., 2004, 126, 14740 CrossRef PubMed.
- J. D. Sunderhaus and S. F. Martin, Chem. – Eur. J., 2009, 15, 1300 CrossRef CAS PubMed; D. S. Tan, Nat. Chem. Biol., 2005, 1, 74 CrossRef PubMed; S. L. Schreiber, Science, 2000, 287, 1964 CrossRef.
- D. Robbins, A. F. Newton, C. Gignoux, J.-C. Legeay, A. Sinclair, M. Rejzek, C. A. Laxon, S. K. Yalamanchili, W. Lewis, M. A. O'Connell and R. A. Stockman, Chem. Sci., 2011, 2, 2232 RSC.
- A. Barthelme, D. Richards, I. R. Mellor and R. A. Stockman, Chem. Commun., 2013, 49, 10507 RSC.
- J.-C. Legeay, W. Lewis and R. A. Stockman, Chem. Commun., 2009, 2207 RSC.
- M. S. Karatholuvhu, A. Sinclair, A. F. Newton, M.-L. Alcaraz, R. A. Stockman and P. L. Fuchs, J. Am. Chem. Soc., 2006, 128, 12656 CrossRef CAS PubMed; R. A. Stockman, Tetrahedron Lett., 2000, 41, 9163 CrossRef.
- S. J. Roe, D. L. Hughes, P. Aggarwal and R. A. Stockman, Synthesis, 2009, 3775 Search PubMed; S. J. Roe and R. A. Stockman, Chem. Commun., 2008, 3432 RSC.
- A. F. Newton, M. Rejzek, M.-L. Alcaraz and R. A. Stockman, Beilstein J. Org. Chem., 2008, 4, 4 CrossRef CAS PubMed; M. Rejzek, R. A. Stockman and D. L. Hughes, Org. Biomol. Chem., 2005, 3, 73 Search PubMed.
- C. Gignoux, A. F. Newton, A. Barthelme, W. Lewis, M.-L. Alcaraz and R. A. Stockman, Org. Biomol. Chem., 2012, 10, 67 Search PubMed; L. G. Arini, P. Szeto, D. L. Hughes and R. A. Stockman, Tetrahedron Lett., 2004, 45, 8371 CrossRef CAS PubMed.
- A. Sinclair, L. G. Arini, M. Rejzek, P. Szeto and R. A. Stockman, Synlett, 2006, 2321 CAS.
- T. E. Storr, S. J. Cully, M. J. Rawling, W. Lewis, D. Hamza, G. Jones and R. A. Stockman, Bioorg. Med. Chem., 2015, 23, 2621–2628 CrossRef CAS PubMed; M. C. McLeod, G. Singh, J. N. Plampin, III, D. Rane, J. L. Wang, V. W. Day and J. Aube, Nat. Chem., 2014, 6, 133 CrossRef PubMed; M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef PubMed.
- Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673 CrossRef CAS PubMed; C. M. Marson, Chem. Soc. Rev., 2011, 40, 5514 RSC; J. H. Meyer and P. A. Bartlett, J. Am. Chem. Soc., 1998, 120, 4600 CrossRef; J. Ding, M. E. Fraser, J. H. Meyer, P. A. Bartlett and M. N. G. James, J. Am. Chem. Soc., 1998, 120, 4610 CrossRef; W. W. Smith and P. A. Bartlett, J. Am. Chem. Soc., 1998, 120, 4622 CrossRef.
- H. Jhoti, G. Williams, D. C. Rees and C. W. Murray, Nat. Rev. Drug Discovery, 2013, 12, 644 CrossRef CAS PubMed; M. Congreve, R. Carr, C. Murray and H. Jhoti, Drug Discovery Today, 2003, 8, 876 CrossRef.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef CAS.
- R. Lazny and A. Nodzewska, Chem. Rev., 2010, 110, 1386 CrossRef CAS PubMed.
- M. G. Banwell, M. J. Coster, N. L. Hungerford, M. J. Garson, S. Su, A. C. Kotze and M. H. G. Munro, Org. Biomol. Chem., 2012, 10, 154 CAS.
- D. B. Collum, Acc. Chem. Res., 1992, 25, 448 CrossRef CAS.
- H. Sun, K. M. Millar, J. Yang, K. Abboud and B. A. Horenstein, Tetrahedron Lett., 2000, 41, 2801 CrossRef CAS.
- S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900 CrossRef CAS PubMed.
- K. Voigtritter, S. Ghorai and B. H. Lipshutz, J. Org. Chem., 2011, 76, 4697 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC.
- L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397 CrossRef CAS.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data, 1H and 13C NMR spectra. CCDC 1045613 and 1045614. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc05070g |
| This journal is © The Royal Society of Chemistry 2015 |