
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-catalyzed direct α-arylation of ethers with azoles†
Arkaitz
Correa
*a,
Béla
Fiser
b and
Enrique
Gómez-Bengoa
b
aDepartment of Organic Chemistry I, University of the Basque Country (UPV/EHU), Joxe Mari Korta R&D Center, Avda. Tolosa 72, 20018 Donostia-San Sebastión, Spain. E-mail: arkaitz.correa@ehu.es
bDepartment of Organic Chemistry I, University of the Basque Country (UPV/EHU), P.O. Box 1072, 20080 Donostia-San Sebastián, Spain
First published on 14th July 2015
Abstract
The direct α-arylation of cyclic and acyclic ethers with azoles has been achieved, which features a novel iron-catalyzed cross-dehydrogenative coupling (CDC) process. This practical oxidative method allowed the efficient C2-alkylation of a variety of (benzo)azoles constituting straightforward access to heterocycles of utmost medicinal significance and highlighting the convenient use of feedstock substrates and iron catalysts. A preliminary mechanism supported by DFT calculations is discussed as well.
Since the end of the last century sustainable development has constituted a matter of genuine concern for our society and scientific community. As a result, “green chemistry” represents one of the key factors for scientists when designing new chemical processes.1 In this respect, the use of ethers such as tetrahydrofuran (THF) and related derivatives as important raw chemicals for the construction of more complex molecules of pharmaceutical interest has recently received a great deal of attention.2 Indeed, direct functionalization of molecules containing C(sp3)–H bonds stands out today as one of the most challenging and relevant areas in modern organic chemistry offering numerous attractive advantages such as the reduction of the reliance on existing functional groups while improving atom economy and energy efficiency.3 The last few years have witnessed a burgeoning of cross-dehydrogenative couplings (CDCs) involving the use of catalytic amounts of first-row transition metals.4 Based on their low-price, ready availability, and environmentally friendly character, iron salts5 constitute potentially ideal catalysts which offer attractive advantages in this particular area of expertise. Despite the impressive achievements, the assembly of new C–C linkages based upon iron-catalyzed C(sp3)–H functionalization events are still rare in the literature.6
Azoles are prevalent key motifs in a myriad of biologically active compounds, agrochemicals and organic functional materials such as liquid crystals and fluorescent dyes.7 Accordingly, C–H functionalization of azoles is an active area of research which provides simple and rapid access to a plethora of valuable functionalized heterocyclic cores. Whereas arylation, alkenylation, alkynylation and amination processes of azole derivatives have been widely explored,8 direct alkylation still represents a challenge.9N-Tosylhydrazones10 and carboxylic acids11 are among the most common coupling partners to perform C2-alkylation reactions of azoles. Nevertheless, the most straightforward and convenient approach involves the use of non-functionalized ethers via the addition of in situ generated α-oxyalkyl radical species to heteroarenes generally referred to as the Minisci reaction.12 Such processes are of prime importance within medicinal chemistry and have been accomplished using both copper13a and photoredox iridium catalysts13b and even under metal-free conditions.13c While these are efficient and elegant procedures, they still suffer from certain limitations such as the restricted use of (benzo)thiazoles (route a), isoquinolines and pyridines (route c) or requiring harsh reaction conditions like using 4.0 equiv. of the oxidant at high temperatures (route b). In this context, we envisioned whether the use of iron salts would facilitate the development of a complementary and advantageous strategy for the C2-alkylation of azoles with a relatively broad scope and operational simplicity. In fact, iron complexes are known to react with alkyl peroxides to generate organic radical species which can further act as powerful oxidizing agents.14 Herein we describe a novel CDC of (benzo)azoles and ethers featuring the efficient use of a combination of FeF2 and organic peroxides as the oxidant (Scheme 1).
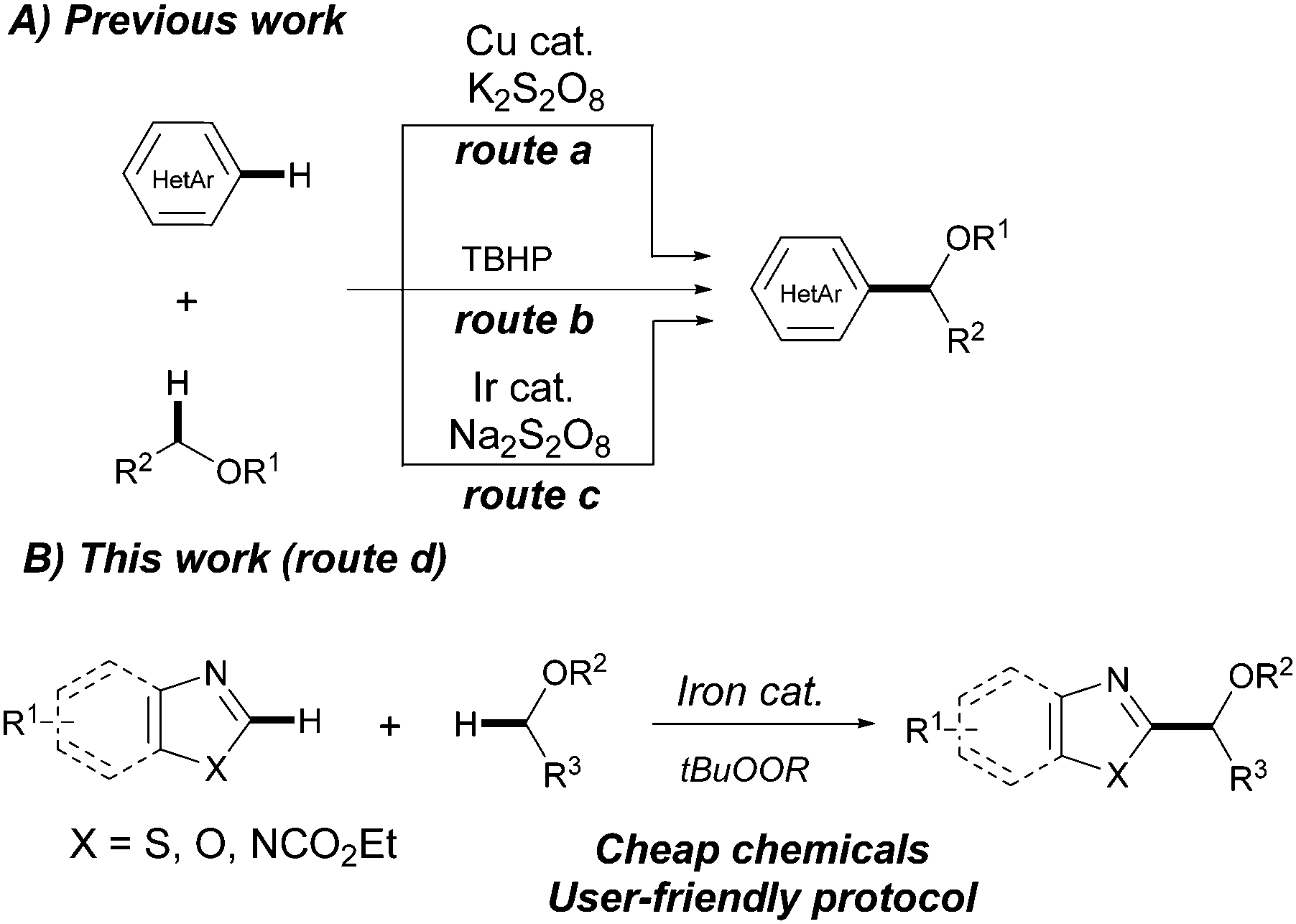 | ||
| Scheme 1 Direct α-arylation of cyclic and acyclic ethers with azoles. | ||
We initially selected the direct coupling of benzothiazole (1a) and tetrahydrofuran (THF, 2a) as the model system to evaluate the feasibility of our approach. We anticipated that the nature of the metal source and the oxidant would have a profound impact on reactivity and accordingly the effect of such variables was systematically examined.15 To our delight, the target CDC event took place in a remarkable 51% yield when utilizing a combination of FeF2 and tert-butyl peroxybenzoate at 90 °C (Table 1, entry 6). Further screening of the oxidants clearly revealed that TBHP was the best choice while other common oxidants were much less effective (Table 1, entries 1–8). It is worth noting that the process was found to be compatible with the use of an aqueous solution of TBHP, albeit the product was obtained in a comparatively low yield (entry 8). Importantly, the catalytic activity was highly dependent on the counteranion and the use of other fluoride salts seemed to have a crucial effect on the reaction outcome. FeF3 was found to be as efficient as FeF2 (entry 12), but other iron sources (entries 9–11) as well as other fluoride metal salts (entries 13 and 14) provided lower yields.15 Remarkably, the yield was dramatically improved upon reducing the amount of THF and adding 1,2-dichloroethane as the co-solvent. Under those conditions the amount of oxidant could be significantly reduced to 1.0 equivalent and 3a was obtained in 80% yield (entry 17). The performance of the process under an air atmosphere was detrimental to the reaction, although 3a was obtained in 62% yield. The addition of other additives or variation of the temperature were found to be ineffective in improving the catalyst performance.15–16 Additionally, several control experiments evidenced that both the iron catalyst and the peroxide were critical for success (Table 1, entries 15 and 16).
|
|
|||
|---|---|---|---|
| Entry | Metal salt | Oxidant | 3a (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (1.0 mL), metal salt (10 mol%), oxidant (2.0 equiv.) at 90 °C for 24 h under argon. b Yield of the isolated product after column chromatography. c TBHP (1.0 equiv.) using 2a (0.5 mL) in 1,2-dichloroethane (0.5 mL). d Under air. e at 80 °C. TBHP = tert-butyl hydroperoxide (5.0–6.0 M in decane); TBHP aq = 70 wt% tBuOOH in H2O. | |||
| 1 | FeF2 | K2S2O8 | 0 |
| 2 | FeF2 | DDQ | 0 |
| 3 | FeF2 | Cumene hydroperoxide | 0 |
| 4 | FeF2 | Dicumyl peroxide | 0 |
| 5 | FeF2 | tBuOOtBu | Traces |
| 6 | FeF2 | tBuOOBz | 51 |
| 7 | FeF2 | TBHP | 62 |
| 8 | FeF2 | TBHP aq | 41 |
| 9 | FeCl2 | TBHP | Traces |
| 10 | Fe(OAc)2 | TBHP | 43 |
| 11 | Fe(acac)3 | TBHP | 38 |
| 12 | FeF3 | TBHP | 61 |
| 13 | CoF2 | TBHP | 47 |
| 14 | CuF2 | TBHP | 29 |
| 15 | None | TBHP | 9 |
| 16 | FeF2 | None | 0 |
| 17 | FeF 2 | TBHP | 80 , (62) |
| 18 | FeF2 | TBHP | 60c,e |

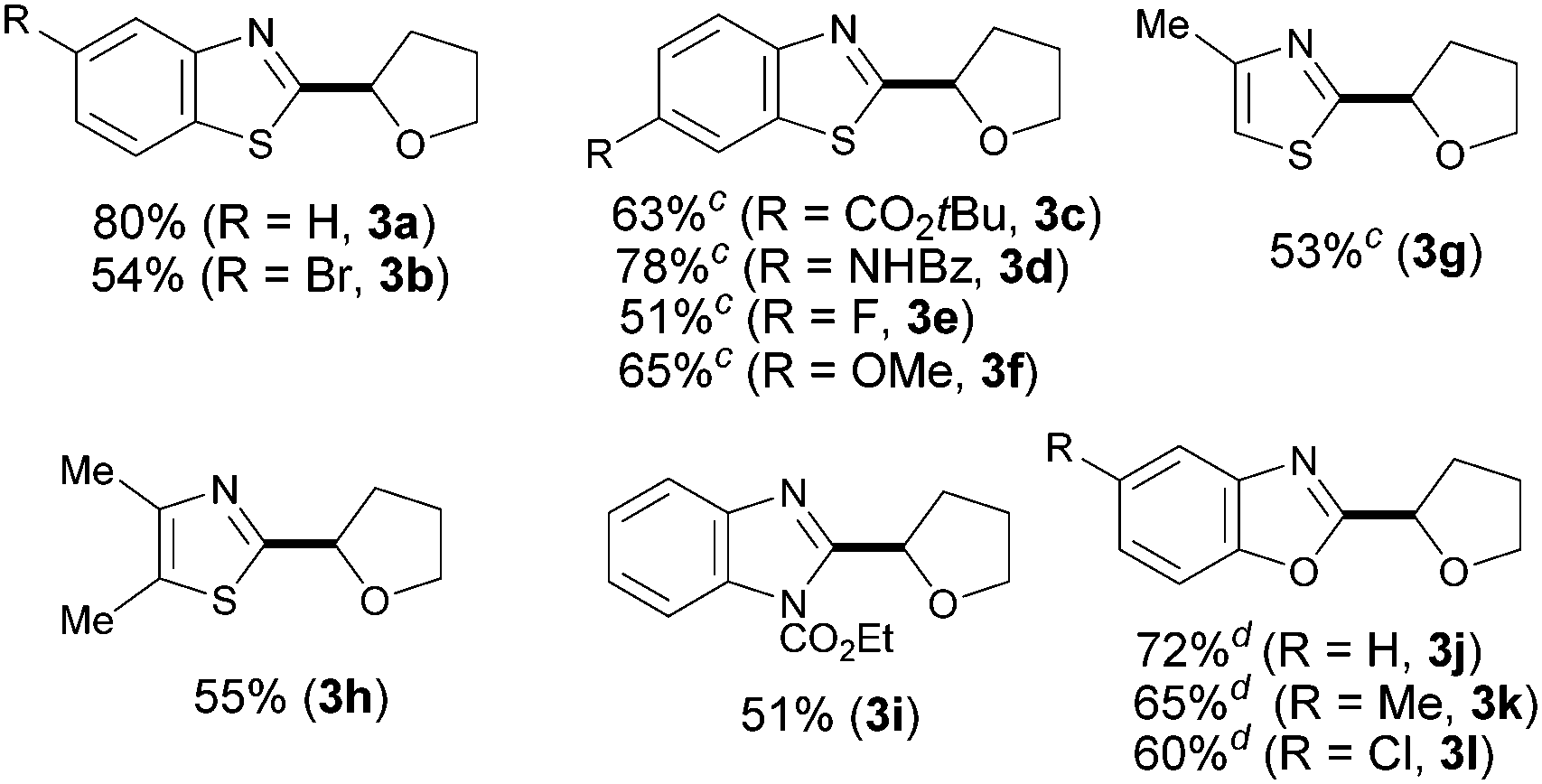
Having identified the optimal reaction conditions, we next focused on examining the preparative scope and generality of our iron-catalyzed direct arylation event. As shown for 3a–f, moderate to good yields were obtained when differently substituted benzothiazoles were utilized. It is noteworthy that electron-deficient derivatives provided lower yields since full conversion was not achieved. Importantly, several functional groups were accommodated such as esters (3c), amides (3d), halides (3b and 3e), and ethers (3f). Strikingly, the strongly coordinating nitrogen motif in 3d did not interfere with the coupling, which reveals a low Lewis acidity, if any, of our catalyst system. Of particular importance is the compatibility with the presence of halides, which provides additional functionalization opportunities via cross-coupling techniques. Notably, the method was found applicable for the preparation of non-benzofused thiazoles (3g–h) and benzimidazoles (3i), albeit the products were obtained in moderate yields. When benzoxazole derivatives were subjected to the optimized conditions, the desired products were not detected. Gratifyingly, minor modifications on the reaction conditions such as replacing the use of TBHP by tert-butyl peroxybenzoate allowed for the efficient coupling of several benzoxazoles (3j–l).17 In these cases, the less basic benzoate species are generated by homolytic cleavage of the oxidant and hence the corresponding coupling product can be satisfactorily obtained,18 a significant improvement compared to the parent Cu-catalyzed process (Table 2).13a
|
|
|---|
a Reaction conditions: 1 (0.5 mmol), FeF2 (10 mol%), TBHP (1.0 equiv., 5.0–6.0 M in decane) in a mixture 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCE (1:1, 1.0 mL) at 90 °C for 24 h under argon.
b Yield of the isolated product after column chromatography, average of at least two independent runs.
c TBHP (2.0 equiv.) using 2a (1.0 mL).
d
tBuOOBz (2.0 equiv.) using 2a (1.0 mL). :DCE (1:1, 1.0 mL) at 90 °C for 24 h under argon.
b Yield of the isolated product after column chromatography, average of at least two independent runs.
c TBHP (2.0 equiv.) using 2a (1.0 mL).
d
tBuOOBz (2.0 equiv.) using 2a (1.0 mL).
|
|
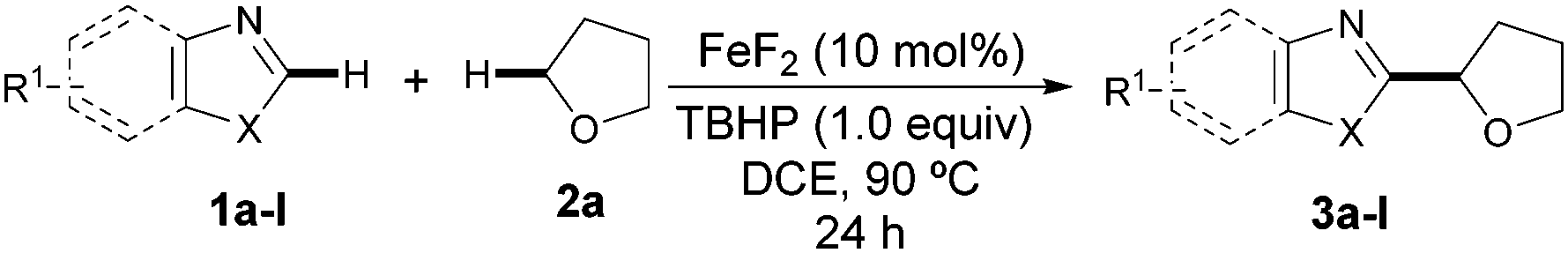
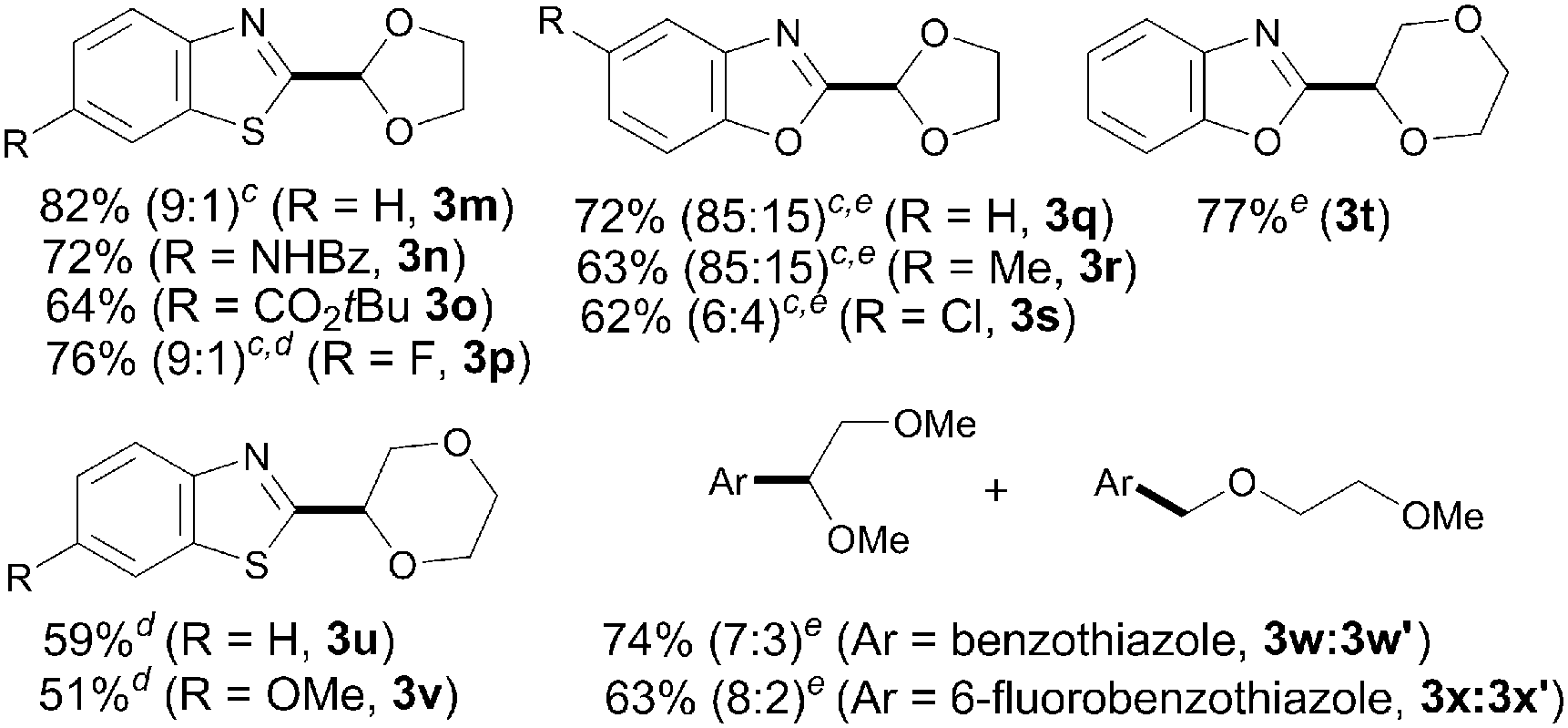
Aside from THF, other related cyclic and acyclic ethers are commonly used as solvents in chemical processes and are prevalent key structures in a wide range of valuable compounds. Of particular interest is 1,3-dioxolane given that its coupling would provide a masked formyl derivative through a practical and aldehyde-free synthetic protocol. Accordingly, we next explored the scope of our iron-catalyzed heteroarylation process regarding the ether coupling partner. As shown in Table 3, a wide variety of differently substituted benzothiazoles and benzoxazoles smoothly underwent the coupling with 1,3-dioxolane to afford the corresponding acetal derivatives in good yields (3m–s). Remarkably, 1,3-dioxolane reacted selectively at the C2 position versus the less reactive C4 atom providing 3n and 3o as single isomers. However, in most cases both isomers were detected with high regioselectivity (up to 9:1); whereas the products 3m and 3p bearing the benzothiazole core were easily separated by column chromatography, the benzoxazole derivatives 3q–3s were isolated as inseparable mixtures of both isomers (regioselectivity up to 85:15 determined by 1H NMR spectroscopy). Interestingly, 1,4-dioxane could also be utilized to furnish the corresponding coupling products in moderate to good yields (3t–v). It is noteworthy that the acyclic ether 1,2-dimethoxyethane also underwent the target reaction at both methylene and methyl sites with good combined yields and high regioselectivities (up to 8:2; 3w:3w′ and 3x:3x′). Unfortunately, other coupling partners such as dibutyl ether and ethanol or less acidic heterocycles such as 1,2,3-triazoles and indoles were found unreactive under our optimized conditions.
|
|
|---|
|
a Reaction conditions: 1 (0.5 mmol), FeF2 (10 mol%), TBHP (1.0 equiv., 5.0–6.0 M in decane) in a mixture 2a:DCE (1:1, 1.0 mL) at 90 °C for 24 h under argon.
b Yield of the isolated product after column chromatography, average of at least two independent runs.
c Ratio of C2 vs. C4 isomer.
d TBHP (2.0 equiv.) using 2 (1.0 mL).
e
tBuOOBz (2.0 equiv.) using 2 (1.0 mL).
|
|
Although the detailed mechanistic picture clearly requires further studies, several control experiments as well as DFT studies15,19 were performed to gain some insights into the reaction mechanism. The CDC event was entirely suppressed upon addition of radical scavengers such as BHT and 1,1-diphenylethylene; interestingly, in the latter case the coupling product 4 was isolated instead, in 10% yield.20 Besides, the addition of TEMPO results in very low conversion of the azole and just traces of the product were detected. These experimental pieces of evidence tentatively support a radical pathway. Notably, subsequent competition experiments with benzoxazole 1j utilizing an equimolecular mixture of THF/THF-d8 showed a significant kinetic isotopic effect (kH/kD = 3.18), thus suggesting that the C(sp3)–H bond cleavage with concomitant formation of an α-oxyalkyl radical is likely the rate-determining step (Scheme 2). In order to clarify the role of the iron catalyst, Sc(OTf)3, Bi(OTf)3 and AlCl3 were used instead and the coupling product 3a was obtained in much lower yields; hence it is unlikely that FeF2 is acting as a simple Lewis acid.15,21 Based on the above results, a plausible mechanism supported by DFT studies is outlined in Scheme 3. Initially, FeF2 facilitates the homolytic cleavage of the starting oxidant to form the hydroxide and tert-butoxy radical species under heating conditions.6b,22 Computational data confirm that the homolytic cleavage of tBuOOH is a highly endergonic process, with an uphill Gibbs Free energy of 5.1 kcal mol−1 and the Fe catalyst helps stabilize the arising radical species by the formation of a very stable Fe(III) complex, which lies ca. 80 kcal mol−1 lower in energy than the starting reactants. Next, the C(sp3)–H adjacent to the oxygen atom of THF can be abstracted by tert-butoxy radical species to furnish I, with an activation energy of only 12.5 kcal mol−1,23 and further oxidized through a SET event to the corresponding oxonium cation II by FeF2(OH), lying ca. 5 kcal mol−1 lower in energy than the sum of the starting Fe(III) complex and radical species.24 Finally, the hydroxide anion is basic enough to easily deprotonate the azole 1a, with a low activation energy of only 2.6 kcal mol−1, which eventually reacts with oxonium ion II through an extremely favorable process (ΔGR = −82.1 Kcal mol−1).25 On balance then, we assume that FeF2 plays a key redox role in assisting both the heterolytic cleavage of the oxidant and the oxidation of the carbon radical I to the oxonium ion II.
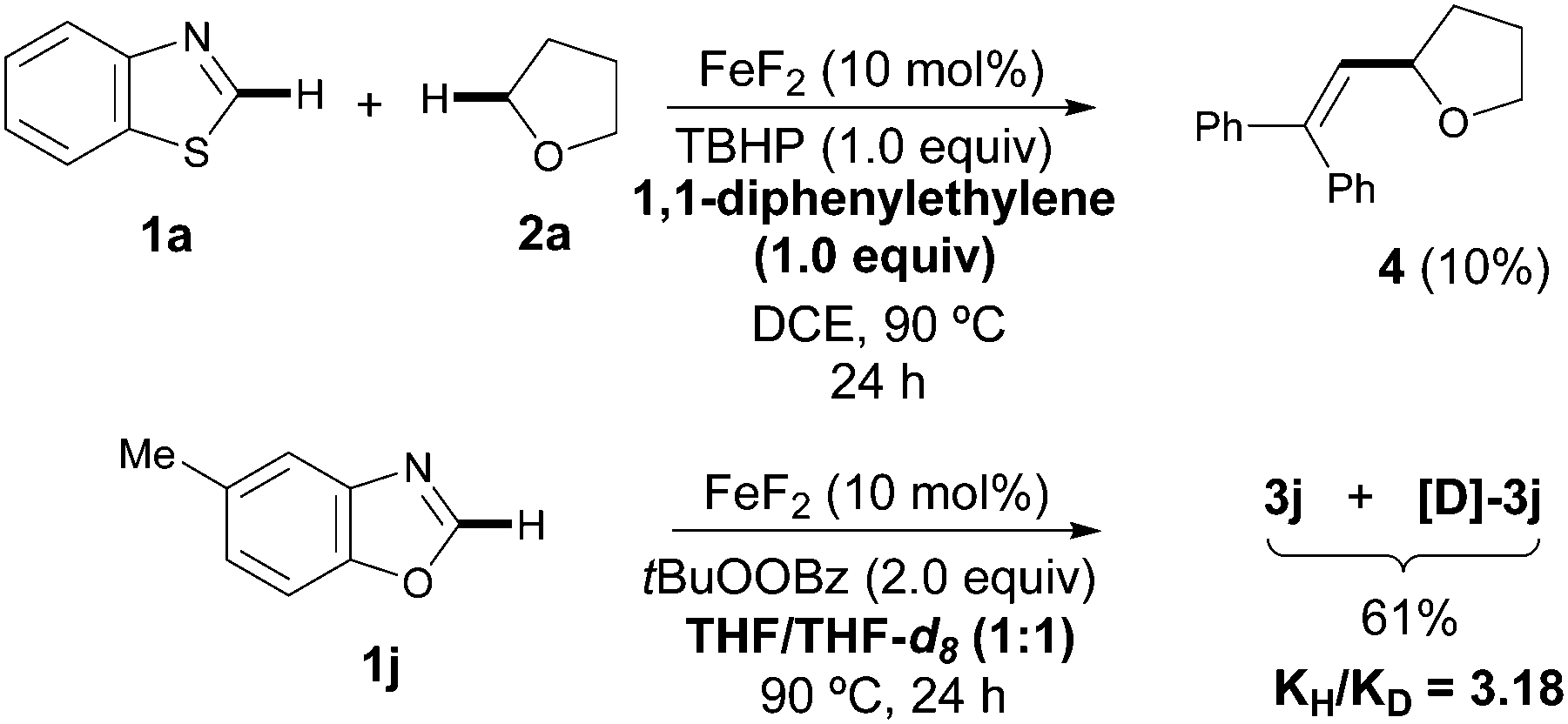 | ||
| Scheme 2 Control experiments. | ||
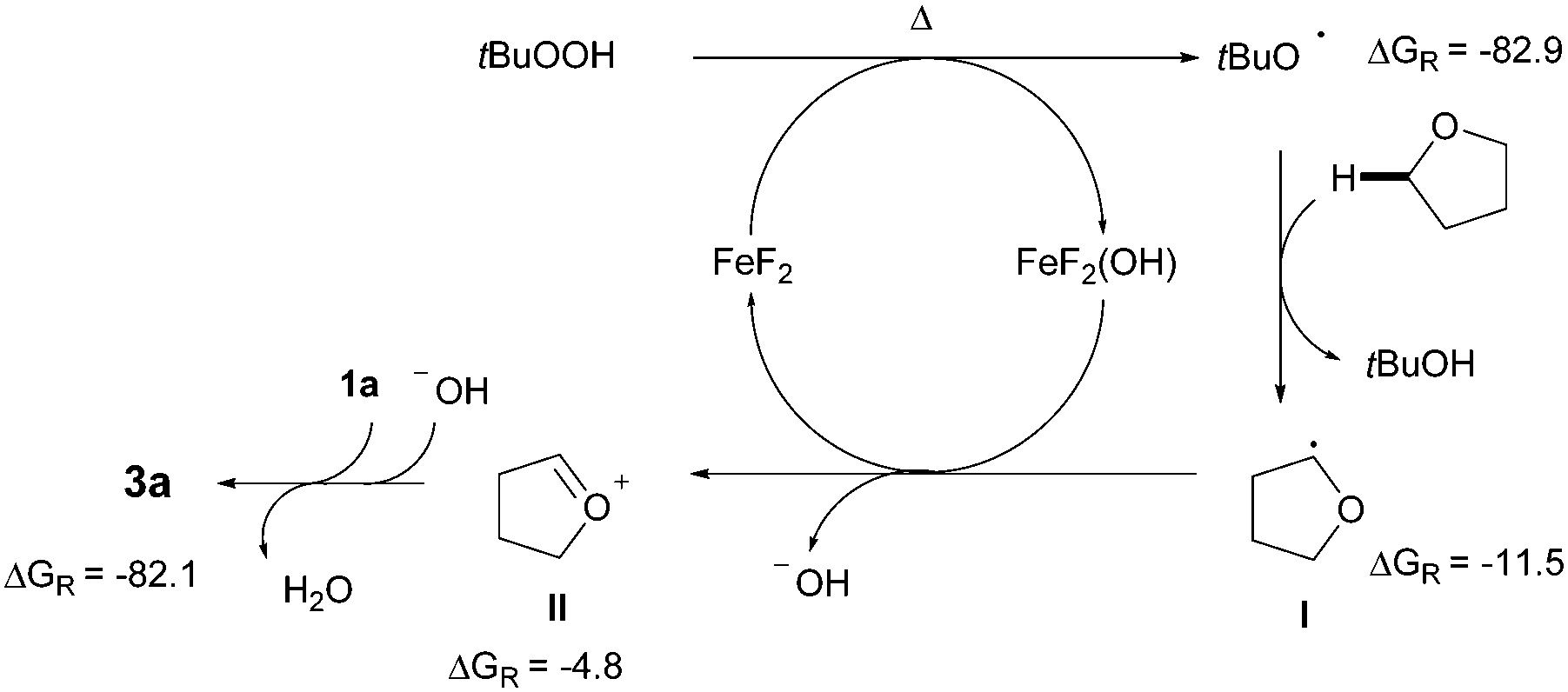 | ||
| Scheme 3 Proposed mechanism. | ||
In summary, we have developed a novel catalytic approach to the direct α-heteroarylation of cyclic and acyclic ethers with azoles. This practical and environmentally friendly protocol highlights the advantageous use of iron salts and cheap feedstock substrates while featuring a dual C–H bond oxidative cross-coupling. Furthermore, the method was found to be applicable to the assembly of a wide variety of functionalized heterocycles of paramount medicinal importance and represents an attractive, yet complementary, strategy for the C–H alkylation of azoles. We anticipate that our experimental and computational studies could lead to acquiring new knowledge in catalyst design, thus opening up new vistas in iron-catalyzed C–H functionalization events.
We thank MINECO for a Ramón y Cajal research contract (RYC-2012-09873) and FP7 Marie Curie Actions of the European Commission via the ITN ECHONET network (MCITN-2012-316379) for financial support. We also thank SCAB from SGIker of UPV/EHU for technical support and allocation of computational resources. Prof. J. M. Aizpurua is kindly acknowledged for providing equipment and laboratory facilities.
Notes and references
- (a) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC; (b) K. Sanderson, Nature, 2011, 469, 18 CrossRef CAS PubMed; (c) R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437 RSC.
- For selected examples, see: (a) Y. Zhang and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 4242 CrossRef CAS PubMed; (b) Z. Li, R. Yu and H. Li, Angew. Chem., Int. Ed., 2008, 47, 7497 CrossRef CAS PubMed; (c) D. Liu, C. Liu, H. Li and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 4453 CrossRef CAS PubMed; (d) W.-T. Wei, M.-B. Zhou, J.-H. Fan, W. Liu, R.-J. Song, Y. Liu, M. Hu, P. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2013, 52, 3638 CrossRef CAS PubMed; (e) M. Wan, Z. Meng, H. Lou and L. Liu, Angew. Chem., Int. Ed., 2014, 53, 13845 CrossRef CAS PubMed; (f) L. Zhou, S. Tang, X. Qi, C. Lin, K. Liu, C. Liu, Y. Lan and A. Lei, Org. Lett., 2014, 16, 3404 CrossRef CAS PubMed.
- (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) Topics in Current Chemistry, C–H Activation, ed. J.-Q. Yu and Z. Shi, Springer-Verlag, Berlin, vol. 292, 2010 Search PubMed; (c) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed.
- For reviews, see: (a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (b) C. J. Scheuermann, Chem. – Asian J., 2010, 5, 436 CrossRef CAS PubMed; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (d) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed . For recent achievements in metal-free CDC events, see: ; (e) X.-F. Wu, J.-L. Gong and X. Qi, Org. Biomol. Chem., 2014, 12, 5807 RSC.
- For general reviews, see: (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS PubMed; (b) A. Fürstner and R. Martin, Chem. Lett., 2005, 34, 624 CrossRef; (c) A. Correa, O. García-Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108 RSC; (d) Topics in Organometallic Chemistry, Iron Catalysis: Fundamentals and Applications, ed. B. Plietker, Springer Verlag, Berlin, vol. 33, 2011 Search PubMed; (e) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed . For early reports on the use of iron salts in the cross-coupling arena, see for example: ; (f) M. Tamura and J. K. Kochi, J. Am. Chem. Soc., 1971, 93, 1487 CrossRef CAS; (g) M. Tamura and J. K. Kochi, J. Organomet. Chem., 1971, 31, 289 CrossRef CAS.
- For a general review, see: (a) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed . For selected papers, see: ; (b) Z. Li, L. Cao and C.-J. Li, Angew. Chem., Int. Ed., 2007, 46, 6505 CrossRef CAS PubMed; (c) Y.-Z. Li, B.-J. Li, X.-Y. Lu, S. Lin and Z.-J. Shi, Angew. Chem., Int. Ed., 2009, 48, 3817 CrossRef CAS PubMed; (d) H. Li, W. Li, W. Liu, Z. He and Z. Li, Angew. Chem., Int. Ed., 2011, 50, 2975 CrossRef CAS PubMed; (e) X.-H. Yang, W.-T. Wei, H.-B. Li, R.-J. Song and J. H. Li, Chem. Commun., 2014, 50, 12867 RSC; (f) S. Murru, C. S. Lott, F. R. Fronczek and R. S. Srivastava, Org. Lett., 2015, 17, 2122 CrossRef CAS PubMed.
- (a) R. E. Martin, L. G. Green, W. Guba, N. Kratochwil and A. Christ, J. Med. Chem., 2007, 50, 6291 CrossRef CAS PubMed; (b) J. Easmon, G. Pürstinger, K.-S. Thies, G. Heinisch and J. Hofmann, J. Med. Chem., 2006, 49, 6343 CrossRef CAS PubMed; (c) I. K. Mangion, B. D. Sherry, J. Yin and F. J. Fleitz, Org. Lett., 2012, 14, 3458 CrossRef CAS PubMed.
- (a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (b) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed; (c) T. W. Lyons and M. S. Sandford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) K. Hirano and M. Miura, Synlett, 2011, 294 CAS; (e) T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2011, 45, 826 CrossRef PubMed.
- For selected examples, see: (a) X. Zhao, G. Wu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 3296 CrossRef CAS PubMed; (b) T. Yao, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 775 CrossRef CAS PubMed.
- (a) J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 7486 CrossRef CAS PubMed; (b) Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560 RSC.
- Selected references: (a) A. Voutchkova, A. Coplin, N. E. Leadbeater and R. H. Crabtree, Chem. Commun., 2008, 6312 RSC; (b) F. Zhang and M. F. Greaney, Angew. Chem., Int. Ed., 2010, 49, 2768–2771 CrossRef CAS PubMed; (c) K. Yang, C. Zhang, P. Wang, Y. Zhang and H. Ge, Chem. – Eur. J., 2014, 20, 7241 CrossRef CAS PubMed.
- M. A. J. Duncton, Med. Chem. Commun., 2011, 2, 1135 RSC.
- (a) Z. Xie, Y. Cai, H. Hu, C. Lin, J. Jiang, Z. Chen, L. Wang and Y. Pan, Org. Lett., 2013, 15, 4600 CrossRef CAS PubMed; (b) J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565 CrossRef CAS PubMed; (c) T. He, L. Yu, L. Zhang, L. Wang and M. Wang, Org. Lett., 2011, 13, 5016 CrossRef CAS PubMed.
- (a) C. Sheu, S. A. Richert, P. Cofre, B. Ross, A. Sobkowiak, D. T. Sawyer and J. R. Kanofsky, J. Am. Chem. Soc., 1990, 112, 1936 CrossRef CAS; (b) D. T. Sawyer, A. Sobkowiak and T. Matsushita, Acc. Chem. Res., 1996, 29, 409 CrossRef CAS; (c) P. Stavropoulos, R. Celenligil-Cetin and A. E. Taper, Acc. Chem. Res., 2001, 34, 745 CrossRef CAS PubMed and references cited therein.
- For more details see the ESI†.
- All attempts to use iodide sources such as tetrabutylammonium iodide, sodium iodide or potassium iodide were unsuccessful. Besides, the addition of supporting ligands such as DMEDA, TMEDA or 1,10-phenanthroline was detrimental for the catalyst performance.
- This distinct reactivity profile of benzothiazole vs. benzoxazole could be attributed to the higher acidity of the latter and its tendency to open up under basic conditions.
- When using tBuOOBz as the oxidant, benzoic acid was obtained as a side-product, which was easily eliminated upon basic workup of the reaction crude prior to chromatographic purification.
- DFT studies were carried out using the M06 functional as implemented in Gaussian 09 and the 6-311++G** basis sets (SDD for Fe).
- Compound 4 was previously isolated in other processes involving the formation of radical species, see: (a) R. P. Pandit and Y. R. Lee, Adv. Synth. Catal., 2014, 356, 3171 CrossRef CAS; (b) Ref. 2c. For selected CDC events between olefins and C(sp3)–H bonds, see: ; (c) K. Cheng, L. Huang and Y. Zhang, Org. Lett., 2009, 11, 2908 CrossRef CAS PubMed; (d) G. Majji, S. Guin, S. K. Rout, A. Behera and B. K. Patel, Chem. Commun., 2014, 50, 12193 RSC; (e) D. Liu, C. Liu, H. Li and A. Lei, Chem. Commun., 2014, 50, 3623 RSC; (f) Y. Zhu and Y. Wei, Chem. Sci., 2014, 5, 2379 RSC.
- J. I. Padrón and V. S. Martín, Top. Organomet. Chem., 2011, 33, 1 CrossRef.
- (a) H. B. Dunford and J. S. Stillman, Coord. Chem. Rev., 1976, 19, 187 CrossRef CAS; (b) L. Regine and L. Marnett, J. Am. Chem. Soc., 1989, 111, 6621 CrossRef; (c) Y. Zhang and C.-J. Li, Eur. J. Org. Chem., 2007, 4654 CrossRef CAS; (d) S. Pan, J. Liu, H. Li, Z. Wang, X. Guo and Z. Li, Org. Lett., 2010, 12, 1932 CrossRef CAS PubMed.
- Radical I will be probably involved in an equilibrium between its free form and a Fe(III)–OtBu complex by combination with the Fe(II) catalyst, thus compromising the availability of the free radical. See ref. 15.
- Computational data confirm the experimentally observed distinct reactivity profile of FeCl2 and FeF2. Whereas FeCl2 could also facilitate the homolytic cleavage of tBuOOH through a favourable process (ΔGR = −111.1 Kcal mol−1), the subsequent oxidation of intermediate I to oxonium ion II remains a rather unfavourable pathway when using FeCl2 (ΔGR = 47.0 Kcal mol−1).
- At this stage the intermediacy of a radical into an azole derivative and subsequent C–C bond formation through termination of such species and intermediate I cannot be entirely ruled out. However, it remains unlikely given the fact that the bis-heteroaryl compound resulting from the corresponding homocoupling was never detected.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc05005g |
| This journal is © The Royal Society of Chemistry 2015 |