
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeat-induced formation of one-dimensional coordination polymers on Au(111): an STM study†
Tuan Anh
Pham
*a,
Fei
Song
a,
Mariza N.
Alberti
b,
Manh-Thuong
Nguyen
c,
Nils
Trapp
b,
Carlo
Thilgen
b,
François
Diederich
b and
Meike
Stöhr
*a
aZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: t.a.pham@rug.nl; m.a.stohr@rug.nl
bLaboratorium für Organische Chemie, ETH Zürich, Vladimir-Prelog-Weg 3, CH-8093 Zürich, Switzerland
cThe Abdus Salam International Centre for Theoretical Physics, Strada Costiera 11, I-34151, Trieste, Italy
First published on 10th August 2015
Abstract
The formation of one-dimensional coordination polymers of cyano-substituted porphyrin derivatives on Au(111) induced by thermal annealing is demonstrated by means of scanning tunnelling microscopy. The polymer is stabilised by an unusual threefold coordination motif mediated between an Au atom and the cyano groups of the porphyrin derivatives.
The recent increase of interest in the field of molecular self-assembly on surfaces has opened new avenues towards the development of various types of low-dimensional molecular architectures.1 Among them, metal-coordinated one- and two-dimensional (1D and 2D) structures adsorbed on metal surfaces have attracted special attention. This is due to the possibility to control both the size and geometry of these architectures by properly selecting the organic ligands with respect to length, shape and functional endgroups together with the chemical nature of the metal centres and substrates.2 Moreover, an advantage of metal–organic coordination bonds, in comparison to other types of non-covalent and covalent bonds, is that they show an adequate balance between stability and lability due to their relative strength and reversibility.3 This offers the possibility to build-up robust 1D/2D structures with a high level of structural perfection,4 which is of utmost importance for the usage of such networks in future molecular electronic devices. To date, most of the reported metal–organic units stabilizing surface-supported molecular coordination networks are based on the combination of organic linkers bearing pyridyl, cyano, hydroxyl or carboxyl endgroups and transition-metal atoms such as Cu, Fe, Co, Ni.5 Up to now, reports on the formation of 1D/2D molecular coordination networks on an Au surface without the addition of transition-metal atoms are very rare.6
Herein, we report a low-temperature scanning tunnelling microscopy (STM) investigation on the formation of 1D coordination polymers on Au(111) induced by thermal annealing under ultrahigh vacuum (UHV) conditions. For this work, we synthesized porphyrin 1 (see ESI†) with two 3,5-di(tert-butyl)phenyl and two 4′-cyanobiphenyl groups attached to cis-meso-positions (Fig. 1a).
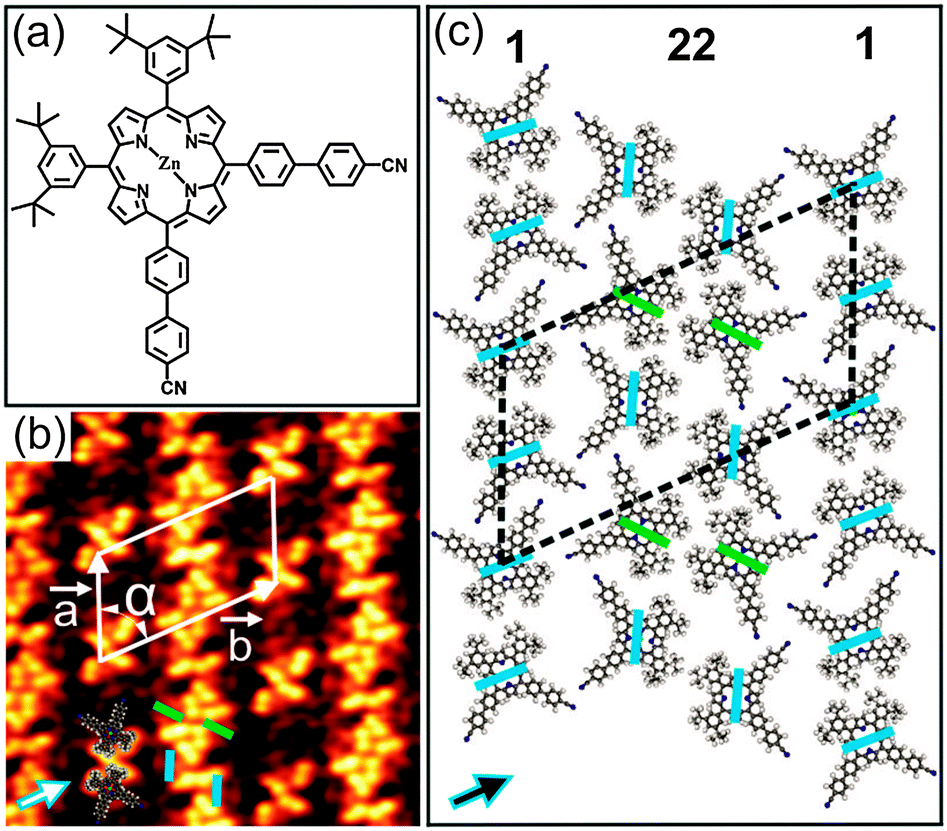 | ||
| Fig. 1 (a) Chemical structure of porphyrin 1. (b) Close-up STM image (15 × 15 nm2, U = −1.8 V, I = 20 pA) showing the 2D close-packed pattern for close to 1 ML coverage. (c) Tentative model for the molecular network. The unit cell vectors a and b are denoted by white arrows in (b). The blue and green bars in (b) represent two different conformational isomers of 1. The arrows in (b) and (c) indicate one of the three principal directions of the substrate. | ||
Upon deposition of submonolayer coverage of 1 on Au(111) held at room temperature (RT), nanoribbon-like structures were observed by STM (Fig. S3, ESI†). Besides, isolated clusters were also found at the elbow sites of the Au(111) herringbone reconstruction which are well-known as preferential nucleation sites.8 Close to monolayer (ML) coverage, a well-ordered close-packed 2D pattern was observed (Fig. 1b and Fig. S4, ESI†). Notably, this 2D pattern is based on the nanoribbon-like structures observed at submonolayer coverage. The high-resolution STM image in Fig. 1b reveals that the nanoribbon-like structures are attached back to back resulting in the close-packed 2D pattern. Interestingly, the co-existence of two different conformational isomers of 1 in the 2D pattern can be clearly discerned (Fig. 1b). The conformational isomers can be identified by the dark bridge, which either separates the 4′-cyanobiphenyl from the 3,5-di(tert-butyl)phenyl substituents (A isomer) or acts as a mirror plane for the molecule (B isomer) (blue and green coloured lines in Fig. 1b and c), respectively. This is similar to our earlier study of 1 adsorbed on Cu(111).7 More information on the conformational isomers can be found in the ESI† (Fig. S5). As determined from STM analysis, the molecules are arranged in a rhombic unit cell with dimensions of a = (43 ± 1.2) Å, b = (70 ± 1.6) Å and α = (60 ± 3)°. The axes of the molecular unit cell are parallel to principal Au directions. The co-existence of three rotational domains having an angle of 120° to each other was observed, reflecting the threefold symmetry of the underlying substrate (Fig. S4b, ESI†). Moreover, the characteristic dark bridges of 1 are aligned along one of the three principal Au directions. These findings point out the significant influence of the underlying substrate on the adsorption behaviour as well as the conformational states of 1 deposited on Au(111). Fig. 1c shows the tentative molecular model for the observed 2D pattern (see also Fig. S6, ESI†). In this model, the molecules are organized in alternating single and twin rows (marked by 1 and 22, respectively), which are parallel to each other. Remarkably, only the A isomer appears in the single rows, whereas both A and B isomers co-exist in the twin rows. The single rows are characterized by the alignment of neighbouring molecules into an antiparallel fashion with an interdigitation of the 4′-cyanobiphenyl units and a close-packing of the tert-butyl groups. In this way, H-bonds are established between the cyano (CN) groups and the phenyl rings as well as van der Waals (vdW) interactions between the tert-butyl groups. On the other hand, the twin rows, where two nanoribbon-like structures meet back to back to form the 2D pattern, are composed of alternating arrangements of A–A and B–B type isomers. Within these twin isomers, the molecules are aligned antiparallel with the tert-butyl groups facing each other (marked by pairs of either blue or green bars in Fig. 1c). Notably, the molecules in the A–A configuration are slightly rotated with respect to the adjacent molecules in the B–B configuration, allowing the CN and C(Ar)–H groups to point toward each other. In this way, the twin rows are stabilised by both CN⋯HC(Ar) H-bonds and vdW interactions between the tert-butyl groups. Similar cyano-aryl hydrogen bonds are established between the single and twin rows. Overall, the close-packed 2D pattern is stabilised by a combination of H-bonds and vdW interactions.
Annealing submonolayer coverage of 1 adsorbed on Au(111) at 160 °C leads to the formation of 1D flexible polymers which exhibit variable lengths (Fig. 2a). Besides, the nanoribbon-like structures were occasionally observed on the surface (Fig. S7, ESI†). Further annealing at 210 °C results in the exclusive formation of 1D flexible polymers (Fig. S8, ESI†). Notably, already at RT, such 1D polymers with shorter lengths were occasionally observed (Fig. S9, ESI†). These observations show that the occurrence of the 1D polymers and the transformation of the nanoribbon-like structures into the 1D polymers depends on the sample and post-annealing temperature, respectively. The close-up STM image in Fig. 2b clearly reveals that the molecules within the 1D polymers do not exhibit preferential orientations with respect to the principal Au directions. Apparently, the 4′-cyanobiphenyl units of two molecules facing each other in an antiparallel way and another 4′-cyanobiphenyl unit of a third molecule point toward each other and form a threefold node. Each molecule connects to two threefold nodes, resulting in the formation of trimers as basic units of the 1D polymers. Interestingly, the 4′-cyanobiphenyl units of 1 in each trimeric unit display an apparent flexibility. This can be associated with a variation of the opening angle of the two cyanobiphenyl units which differs from the intrinsic opening angle of 90° (Fig. 3a and c). Previously, a similar observation was made for 1 adsorbed on Cu(111) upon coordination to Cu adatoms.7 According to that study, the formation of a metal coordination bond is based on supplying additional energy to the system by thermal annealing which enables the lateral distortion of the 4′-cyanobiphenyl legs. This finding is in good agreement with the dependence of the structural transformation on the annealing temperature observed in the present work. Herein, the thermal energy at RT is insufficient for the bending of the 4′-cyanobiphenyl legs, resulting in the formation of mostly nanoribbon-like structures. In contrast, at elevated temperatures, the energy gain is high enough to vary the opening angle of the 4′-cyanobiphenyl legs to coordinate to Au atoms enabling the formation of flexible 1D coordination polymers. We conclude that the 1D polymer chains are stabilised by a threefold coordination motif mediated by the nitrogen lone-pair electrons of the three 4′-cyanobiphenyl legs and one Au atom positioned at the centre. It should be noted that there are two possibilities for the formation of the CN⋯Au coordination motif depending on whether the Au centres are Au adatoms or Au surface atoms. Our density functional theory (DFT) calculations indicate that Au adatoms are involved in the threefold coordination motif (see Fig. S14, ESI†). It was recently reported that the creation of Au adatoms on Au(111) leads to the reordering of the herringbone reconstruction.9 However, in our case, the herringbone reconstruction is completely unaffected upon the 1D polymer formation. Thus, we suggest that the adatoms result from the lattice gas which forms by the continuous attachment/detachment of atoms from the step edges.12
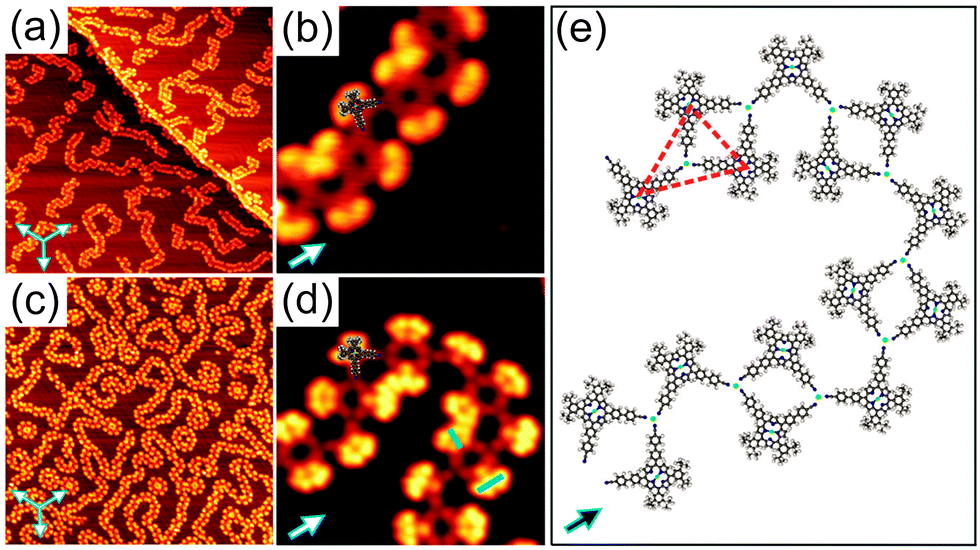 | ||
| Fig. 2 (a and c) Overview STM images (150 × 150 nm2), showing the formation of 1D coordination polymers of 1 on Au(111) induced by annealing at 160 °C and deposition of Co, respectively. The set of three arrows indicates the principal directions of the underlying substrate. (b and d) Close-up STM images (15 × 15 nm2) for (a) and (c), respectively. Blue and green bars in (d) indicate A and B isomers, respectively. (e) The proposed model for the 1D coordination polymer of 1 on Au(111). The green dots in (e) represent Au atoms, while the red dashed triangle illustrates a basic unit of the 1D polymer. The STM images were taken at 77 K with U = −1.8 V, I = 20 pA. | ||
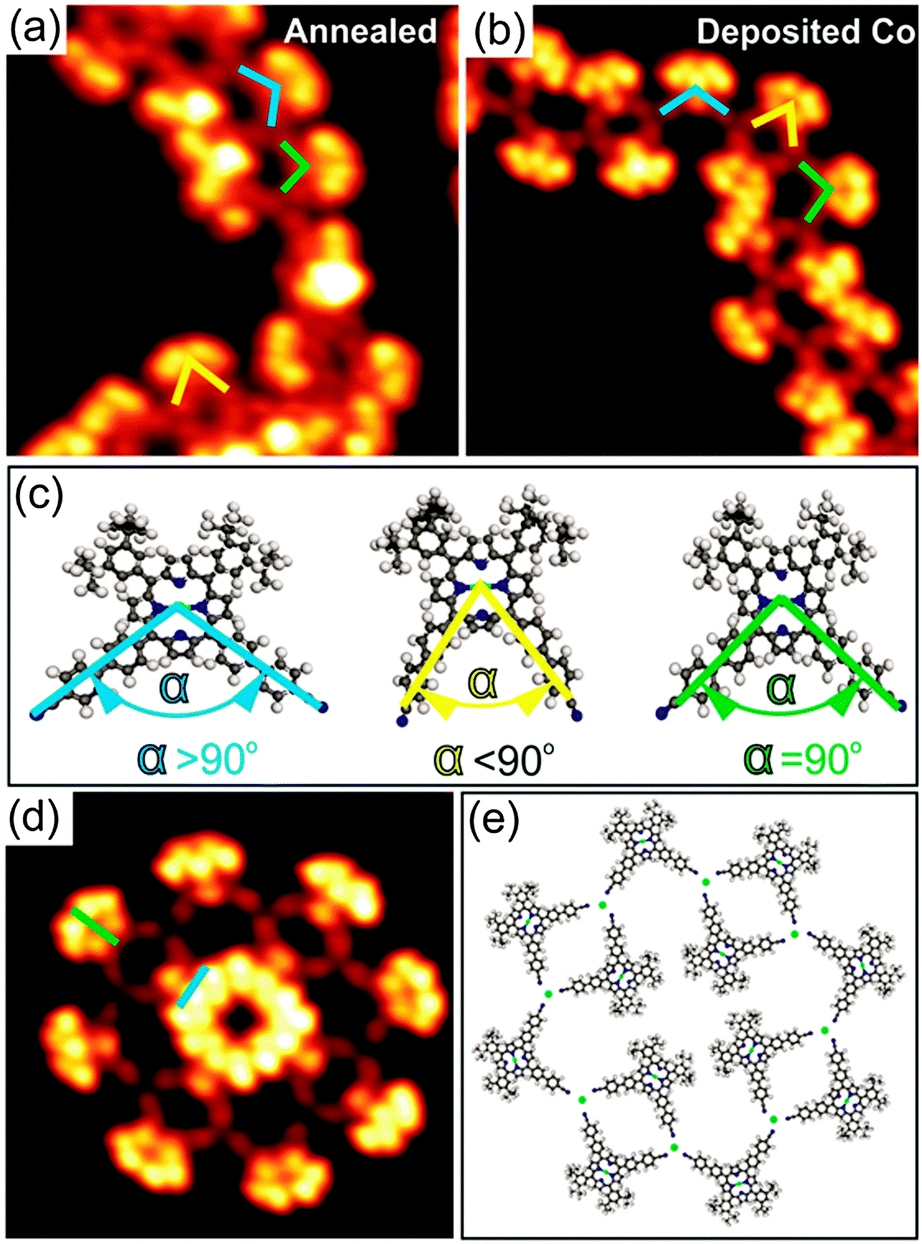 | ||
| Fig. 3 (a and b) Close-up STM images (15 × 15 nm2), showing the lateral distortion of the cyano legs of 1 on Au(111) induced by annealing at 210 °C and deposition of Co, respectively. (c) The coloured lines indicate different angles α defined by the cyano legs of different molecules. Blue: α > 90°; green: α = 90°; yellow: α < 90°. (d) Close-up STM image (10 × 10 nm2) showing one of the two rosette structures observed upon Co deposition and (e) corresponding molecular model. Blue and green bars in (d) indicate A and B isomers, respectively. The green dots in (e) represent Co atoms. The STM images were taken at 77 K with U = −1.8 V, I = 20 pA. | ||
To shed light on the formation of the 1D coordination polymers on Au(111), we performed a control experiment. Cobalt (Co) atoms are commonly used as metal centres for the coordination to CN ligands in the construction of two-dimensional metal–organic frameworks on metal surfaces based on a threefold coordination motif.4a,10 With this knowledge in mind, we deposited Co atoms onto submonolayer coverage of 1 on Au(111) held at RT. The sample was then cooled down to 77 K prior to STM measurements without any additional annealing treatment. Again, flexible 1D polymers were observed as it is the case for annealing of 1 on Au(111) at 160 °C (Fig. 2c). The high-resolution STM image (Fig. 2d) unambiguously shows that the binding motif stabilizing the 1D chains is the same as observed for 1 on Au(111) upon annealing (see also Fig. S10, ESI†). As expected, three N atoms of the 4′-cyanobiphenyl groups point toward one Co atom to form the threefold coordination motif, which is responsible for the 1D polymer formation. Within the 1D polymers both conformational isomers can be clearly discerned (marked by blue and green bars in Fig. 2d). They are randomly distributed within the 1D coordination polymer with equal amounts of each as determined from a statistical analysis. Importantly, the lateral distortion of the 4′-cyanobiphenyl legs, imposed by the coordination to the Co centres, was again observed (Fig. 3b and c). In addition to the 1D polymers, rosette structures are formed upon addition of Co, which were not observed in the annealing case. Rosettes of two different sizes are present (Fig. 3d and Fig. S11, ESI†) and they are stabilised by the same coordination motif as in the 1D coordination polymers (Fig. 3e). Notably, the dark bridges due to the saddle shape conformation of 1 do not align along the principal Au directions. That is in contrast to what we observed for the 2D close-packed structure. From this observation we conclude that the molecule–substrate interaction is mainly mediated by the Co atoms, and the porphyrin core–Au surface interaction is only of weak physisorptive nature.11 Putting it differently, a dominant CN⋯Co coordination interaction is thought to allow for different rotational alignments of 1 with respect to the principal Au directions, thus enabling the formation of rosette structures. Other types of rosette structures have been found recently for the coordination of Fe atoms to pyridyl-substituted porphyrins on Au(111).5d Based on the results obtained from the control experiment, we conclude that the 1D polymers formed upon annealing submonolayer coverage of 1 on Au(111) are indeed stabilised by a threefold CN⋯Au coordination motif. The molecular model for the heat-induced 1D coordination polymers is shown in Fig. 2e.
Another difference between the Au- and Co-coordinated structures can be observed upon studying the molecular arrangement at the Au(111) step edges. Fig. 4a and c show overview STM images of samples prepared by annealing and Co deposition, respectively, exhibiting several step edges. In both cases, the step edges are almost completely covered by molecules. The close-up STM images (Fig. 4b and d) reveal a significant difference in the molecular arrangement at the step edges for the two cases. Upon annealing at 210 °C, the molecules have a preferred orientation with their tert-butyl groups aligned along the step edges. Now, four CN-groups of four neighbouring molecules point toward each other. They meet in a fourfold node, resulting in the formation of a square-like tetramer which is considered as a basic unit of the 1D molecular chains aligned along the step edges (see also Fig. S12, ESI†). In this configuration, the formation of such fourfold nodes is very unlikely for uncoordinated CN-groups due to the electrostatic repulsion of the nitrogen lone-pair electrons. Thus, we suggest that a fourfold Au–CN coordination motif stabilises the 1D chains aligned along the step edges. This observation is in line with recent reports on other CN-substituted molecules adsorbed on Au(111).6b,d Notably, a lateral distortion of the CN-legs with an opening angle larger than 90° due to the coordination bonding was observed (see molecular model in Fig. 4b). In contrast, no such fourfold coordination motifs were observed at the step edges upon Co deposition (Fig. 4d). Importantly, the fourfold CN⋯Au coordination motif not only appears at the Au(111) step edges upon annealing, but was also occasionally found to co-exist with the threefold binding motif in the 1D polymers even at RT (Fig. S9e, ESI†), indicating the flexibility of the Au–CN coordination motif.
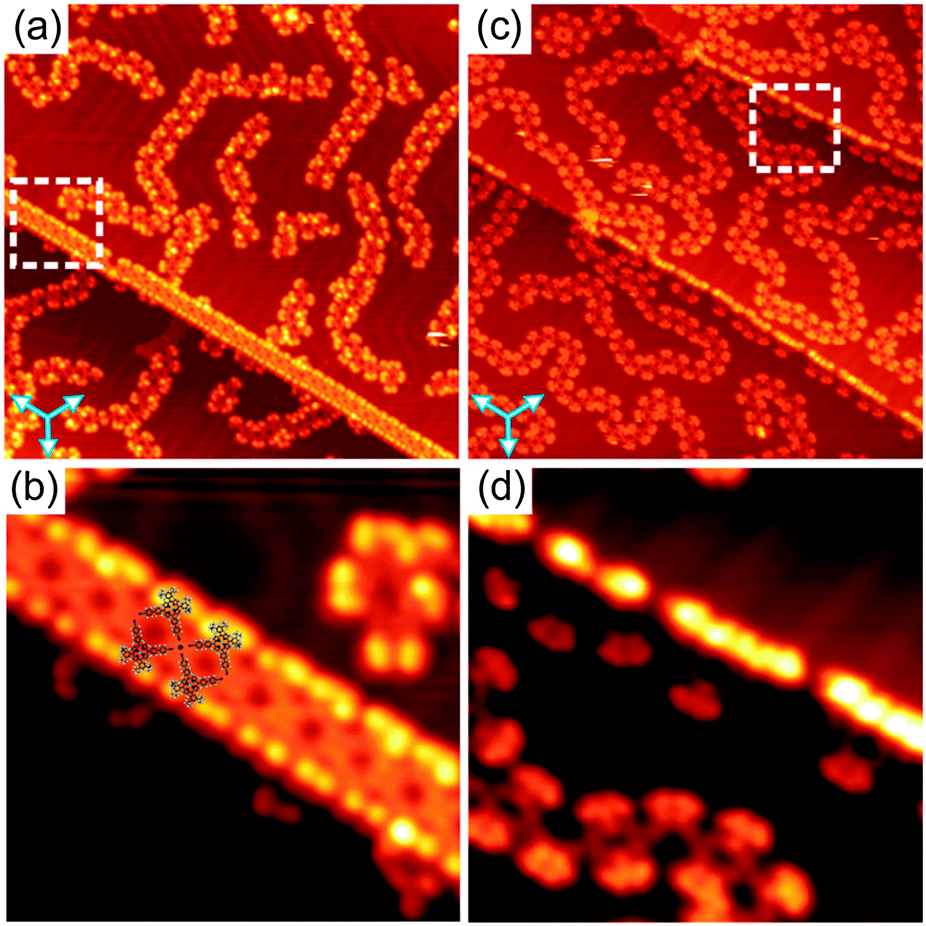 | ||
| Fig. 4 (a and c) Overview STM images (100 × 100 nm2) showing the molecular arrangement of 1 at the step edges of the Au(111) surface after annealing at 210 °C and Co deposition, respectively. The set of three arrows indicates the principal directions of the underlying substrate. (b and d) Close-up STM images (20 × 20 nm2) of the areas marked by white dashed squares in (a) and (c), respectively. In (b) four molecular models are shown to illustrate the fourfold coordination motif. The STM images were taken at 77 K with U = −1.8 V, I = 20 pA. | ||
For coverages close to 1 ML, we also carried out STM measurements with the same experimental protocols (annealing at 160 °C vs. deposition of Co atoms). As a result, a densely packed network of 1D polymers based on the threefold coordination motif was formed on Au(111) after thermal annealing as well as Co deposition (Fig. S13, ESI†). The increase in coverage did not have an influence on the observed types of intermolecular interactions.
In conclusion, for 1 adsorbed on Au(111) we observed the heat-induced formation of flexible 1D coordination polymers stabilised by an unusual threefold coordination motif which is based on the interaction of one Au surface atom with the N atoms of three terminal CN-groups. This finding is strongly supported by the formation of similar 1D coordination polymers with the same binding motif upon Co deposition onto 1 adsorbed on Au(111). In addition, a fourfold CN⋯Au coordination motif was found at the Au(111) step edges upon annealing. Using vicinal Au surfaces could be an effective way to exclusively form straight 1D polymers based on the fourfold CN⋯Au coordination motif, while the addition of Co atoms only results in the formation of threefold coordination motifs. In this way, the desired kind of coordination structures can be easily steered.
T.A.P., F.S., and M.S. acknowledge financial support from the Netherlands Organization for Scientific Research (NWO, Chemical Sciences, VIDI-grant No. 700.10.424) and the European Research Council (ERC-2012-StG 307760-SURFPRO). Research at the ETH Zürich was supported by the Swiss National Science Foundation (SNF). The research of M.N.A. was carried out within the framework of the action “Supporting Postdoctoral Researchers” of the Operational Program “Education and Lifelong Learning” (action's beneficiary: General Secretariat for Research and Technology), and was co-financed by the European Social Fund (ESF) and the Greek State. The authors thank Mr Michael Solar for collecting X-ray data.
Notes and references
- (a) J. A. A. W. Elemans, S. Lei and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7298 CrossRef CAS PubMed; (b) L. Bartels, Nat. Chem., 2010, 2, 87 CrossRef CAS PubMed; (c) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed; (d) M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619 RSC; (e) M. J. Kory, M. Wörle, T. Weber, P. Payamyar, S. W. Van de Poll, J. Dshemuchadse, N. Trapp and A. D. Schlüter, Nat. Chem., 2014, 6, 779 CrossRef CAS PubMed.
- (a) H. Spillmann, A. Dmitriev, N. Lin, P. Messina, J. V. Barth and K. Kern, J. Am. Chem. Soc., 2003, 125, 10725 CrossRef CAS PubMed; (b) N. Lin, S. Stepanow, M. Ruben and J. V. Barth, Top. Curr. Chem., 2009, 287, 1 CrossRef CAS; (c) Z. Wang, J. Liu, B. Lukose, Z. Gu, P. G. Weidler, H. Gliemann, T. Heine and C. Wöll, Nano Lett., 2014, 14, 1526 CrossRef CAS PubMed; (d) H. Walch, J. Dienstmaier, G. Eder, R. Gutzler, S. Schlögl, T. Sirtl, K. Das, M. Schmittel and M. Lackinger, J. Am. Chem. Soc., 2011, 133, 7909 CrossRef CAS PubMed; (e) A. Shchyrba, C. Wäckerlin, J. Nowakowski, S. Nowakowska, J. Björk, S. Fatayer, J. Girovsky, T. Nijs, S. C. Martens, A. Kleibert, M. Stöhr, N. Ballav, T. A. Jung and L. H. Gade, J. Am. Chem. Soc., 2014, 136, 9355 CrossRef CAS PubMed.
- (a) C.-C. You, R. Dobrawa, C. R. Saha-Möller and F. Würthner, Top. Curr. Chem., 2005, 258, 39 CrossRef CAS; (b) J. V. Barth, Surf. Sci., 2009, 603, 1533 CrossRef CAS PubMed.
- (a) D. Kühne, F. Klappenberger, R. Decker, U. Schlickum, H. Brune, S. Klyatskaya, M. Ruben and J. V. Barth, J. Am. Chem. Soc., 2009, 131, 3881 CrossRef PubMed; (b) T. Lin, X. S. Shang, J. Adisoejoso, P. N. Liu and N. Lin, J. Am. Chem. Soc., 2013, 135, 3576 CrossRef CAS PubMed.
- (a) G. Pawin, K. L. Wong, D. Kim, D. Sun, L. Bartels, S. Hong, T. S. Rahman, R. Carp and M. Marsella, Angew. Chem., Int. Ed., 2008, 47, 8442 CrossRef CAS PubMed; (b) S. L. Tait, Y. Wang, G. Costantini, N. Lin, A. Baraldi, F. Esch, L. Petaccia, S. Lizzit and K. Kern, J. Am. Chem. Soc., 2008, 130, 2108 CrossRef CAS PubMed; (c) J. Björk, M. Matena, M. D. Dyer, M. Enache, J. Lobo-Checa, L. H. Gade, T. A. Jung, M. Stöhr and M. Persson, Phys. Chem. Chem. Phys., 2010, 12, 8815 RSC; (d) X. F. Mao, T. Lin, J. Adisoejoso, Z. Shi, X. S. Shang, P. N. Liu and N. Lin, Phys. Chem. Chem. Phys., 2013, 15, 12447 RSC; (e) M. E. Garah, A. Ciesielski, N. Marets, V. Bulach, M. W. Hosseini and P. Samorì, Chem. Commun., 2014, 50, 12250 RSC; (f) F. Bebensee, K. Svane, C. Bombis, F. Masini, S. Klyatskaya, F. Besenbacher, M. Ruben, B. Hammer and T. R. Linderoth, Angew. Chem., Int. Ed., 2014, 53, 12955 CrossRef CAS PubMed.
- (a) Z. Shi and N. Li, J. Am. Chem. Soc., 2009, 131, 5376 CrossRef CAS PubMed; (b) M. N. Faraggi, N. Jiang, N. Gonzalez-Lakunza, A. Langner, S. Stepanow, K. Kern and A. Arnau, J. Phys. Chem. C, 2012, 116, 24558 CrossRef CAS; (c) S. Gottardi, K. Müller, J. C. Moreno-Lopez, H. Yildirim, U. Meinhardt, M. Kivala, A. Kara and M. Stöhr, Adv. Mater. Interfaces, 2014, 1, 1300025 Search PubMed; (d) Z. Yang, M. Corso, R. Robles, C. Lotze, R. Fitzner, E. Mena-Osteritz, P. Bäuerle, K. J. Franke and J. I. Pascual, ACS Nano, 2014, 8, 10715 CrossRef CAS PubMed.
- L. A. Fendt, M. Stöhr, N. Wintjes, M. Enache, T. A. Jung and F. Diederich, Chem. – Eur. J., 2009, 15, 11139 CrossRef CAS PubMed.
- (a) D. D. Chambliss, R. J. Wilson and S. Chiang, Phys. Rev. Lett., 1991, 66, 1721 CrossRef CAS; (b) M. Böhringer, K. Morgenstern, W.-D. Schneider, M. Wühn, C. Wöll and R. Berndt, Surf. Sci., 2000, 444, 199 CrossRef.
- (a) A. Saywell, W. Greń, G. Franc, A. Gourdon, X. Bouju and L. Grill, J. Phys. Chem. C, 2014, 118, 1719 CrossRef CAS; (b) H. Zhang, J.-H. Franke, D. Zhong, Y. Li, A. Timmer, O. D. Arado, H. Mönig, H. Wang, L. Chi, Z. Wang, K. Müllen and H. Fuchs, Small, 2014, 10, 1361 CrossRef CAS PubMed.
- (a) U. Schlickum, R. Decker, F. Klappenberger, G. Zoppellaro, S. Klyatskaya, M. Ruben, I. Silanes, A. Arnau, K. Kern, H. Brune and J. V. Barth, Nano Lett., 2007, 7, 3813 CrossRef CAS PubMed; (b) D. Nieckarz and P. Szabelski, J. Phys. Chem. C, 2013, 117, 11229 CrossRef CAS.
- M. Matena, J. Björk, M. Wahl, T.-L. Lee, J. Zegenhagen, L. H. Gade, T. A. Jung, M. Persson and M. Stöhr, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 125408 CrossRef.
- (a) R. C. Jaklevic and L. Elie, Phys. Rev. Lett., 1988, 60, 120 CrossRef CAS; (b) Y. Pan, B. Yang, C. Hulot, S. Blechert, N. Nilius and H.-J. Freund, Phys. Chem. Chem. Phys., 2012, 14, 10987 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of porphyrin 1. Single crystal X-ray analysis of porphyrin 1. Additional STM images. DFT computational details. Copies of 1H, 13C, COSY, HSQC, and HMBC NMR spectra. CCDC 1056340. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04940g |
| This journal is © The Royal Society of Chemistry 2015 |