
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic elucidation of C–H oxidation by electron rich non-heme iron(IV)–oxo at room temperature†
Sujoy
Rana
,
Aniruddha
Dey
and
Debabrata
Maiti
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India
First published on 4th August 2015
Abstract
Non-heme iron(IV)–oxo species form iron(III) intermediates during hydrogen atom abstraction (HAA) from the C–H bond. While synthesizing a room temperature stable, electron rich, non-heme iron(IV)–oxo compound, we obtained iron(III)–hydroxide, iron(III)–alkoxide and hydroxylated-substrate-bound iron(II) as the detectable intermediates. The present study revealed that a radical rebound pathway was operative for benzylic C–H oxidation of ethylbenzene and cumene. A dissociative pathway for cyclohexane oxidation was established based on UV-vis and radical trap experiments. Interestingly, experimental evidence including O-18 labeling and mechanistic study suggested an electron transfer mechanism to be operative during C–H oxidation of alcohols (e.g. benzyl alcohol and cyclobutanol). The present report, therefore, unveils non-heme iron(IV)–oxo promoted substrate-dependent C–H oxidation pathways which are of synthetic as well as biological significance.
High-valent iron–oxo species are responsible for C–H oxidations in numerous biological and chemical transformations for both heme and non-heme enzymes.1 Heme enzymes like cytochrome P450 carry out alkane hydroxylation, olefin epoxidation and sulfoxidation involving the iron(IV)–oxo porphyrin π-cation radical.2 Non-heme enzymes, such as Rieske oxygenase, and α-ketoglutarate dependent dioxygenases, TauD-J, routinely perform biochemical oxidative transformations involving the iron(IV)–oxo intermediate. Intense experimental work has been devoted for mimicking the chemistry of heme/non-heme enzymes.3
Non-heme iron(IV/V)–oxo complexes abstract hydrogen atoms from C–H bonds in the rate determining step to form iron(III) hydroxide and radical species (R˙).4 These active species, depending on their properties, can pursue a radical rebound, radical non-rebound or electron transfer mechanism to form the respective C–H oxidation products (Scheme 1).4l,5 Following a radical rebound pathway, the in situ formed iron(II)-species and alcohol can undergo the comproportionation reaction in the presence of another equivalent of iron(IV)–oxo (Scheme 1).3b In the case of a dissociative pathway, iron(III)–hydroxide and the substrate radical (generated upon HAA) become separated from the solvent cage resulting in subsequent radical trapped products and other side reactions of iron(III)–hydroxides. Such a pathway is well accepted for the iron(IV/V)–oxo and manganese(IV)–oxo complexes.5,6
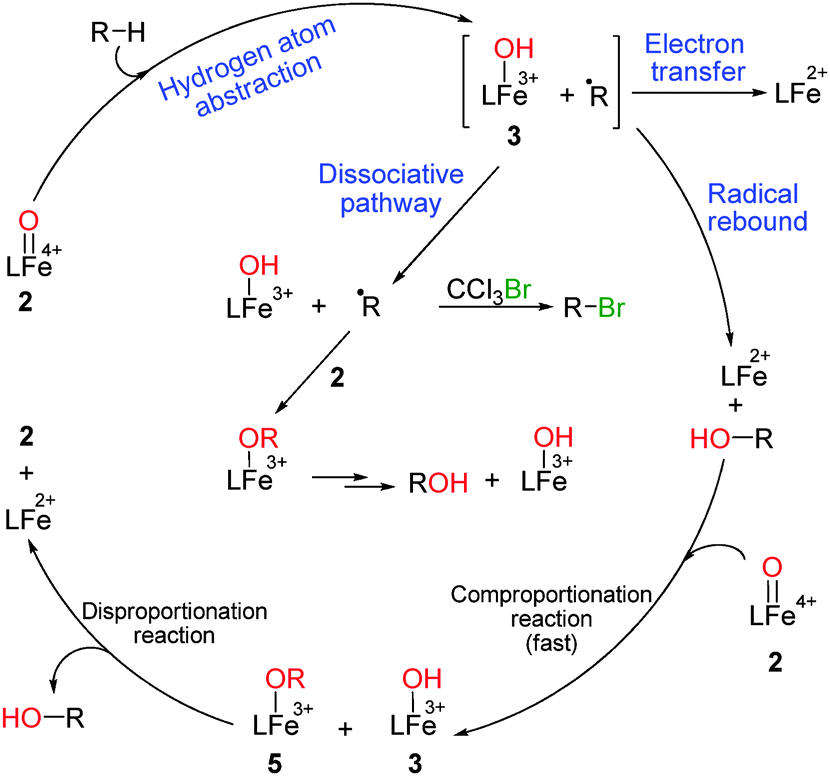 | ||
| Scheme 1 C–H oxidations by non-heme iron(IV)–oxo. | ||
Although the radical rebound pathway has been established for ruthenium(IV)–oxo,7 gathering concrete evidence for the same in the case of iron(IV)–oxo requires further study. We thought of synthesizing a modified N4Py ligand scaffold (L) with electron rich substituents at the picolyl moiety. We were particularly intrigued by the DFT data of Fe–(N4Py) complex which showed greater HOMO contribution by two picolyl moieties that resulted in a shorter Fe–N(picolyl) distance in the Fe–(N4Py) complex.8 We rationalized that the introduction of an electron donating group (such as 4-OMe) in the picolyl unit would further shorten the Fe–(N4Py) distance and would increase the HOMO contribution (Fig. 1).8 Consequently, it would produce more reactive reaction intermediates, which could be verified by detailed mechanistic studies.3d–f,4a,b
 | ||
| Fig. 1 ORTEP diagram of complex 1 (CCDC 1051845) and DFT optimized geometry of 2 using B3LYP/LANL2DZ with N4PyOMe,Me ligand. | ||
The non-heme iron complex [(N4Py)OMe,MeFeII(CH3CN)] (OTf)2 (1) was synthesized by reacting Fe(OTf)2·2CH3CN with an electronically enriched and substituted N4PyOMe,Me ligand.5 Complex 1 was also characterized using X-ray spectroscopy (Fig. 1), ESI-MS (m/z, 688.150), UV-vis spectroscopy (maximum at 459 nm due to LMCT).9 NMR (0–10 ppm, 1H- and 0–200 ppm, 13C-) and EPR (silent) studies indicated the diamagnetic character of 1.10,11 Electrochemical studies of complex 1 showed a lower FeIII/FeII reduction potential (E1/2 ∼ 0.84 V vs. SCE) compared to that of the unsubstituted N4Py iron(II) complex (E1/2 ∼ 1.01 V vs. SCE).8,12 This further suggested that iron(III) species for 1 is more stable compared to that of the unsubstituted N4Py iron(III) complex. The corresponding iron(IV)–oxo species, [(N4Py)OMe,MeFeIV(O)]2+ (2) was synthesized by reacting 1 with iodosyl benzene in acetonitrile at room temperature. A characteristic UV-vis maximum at 692 nm (ε ∼ 432 M−1 cm−1) due to ligand field transitions (d–d transition) was also observed.4a,13 Complex 2 showed a slightly more negative FeIV/FeIII reduction potential (Ep,c ∼ −0.19 V vs. SCE) compared to the unsubstituted [(N4Py)FeIV(O)]2+ (Ep,c ∼ −0.15 V vs. SCE). Notably, 2 was found to be stable at room temperature for a few days (t1/2 ∼ 50 h at 30 °C in air).8 The ESI-MS characterization of complex 2 revealed a major isotopic peak at 704.145 due to [(N4Py)OMe,MeFeIV(O)](OTf)+ which was shifted to 706.150 upon O-18 labeling with H218O (∼95% O-18 incorporation, vide infra).14 The 1H NMR spectrum (−20 to 50 ppm) along with the EPR silent behaviour at 77 K suggested a paramagnetic character of 2 (likely in the S = 1 spin state).10,15
Oxidation of benzyl alcohol by 2 provided benzaldehyde as the sole product (yield, 86%). Labeling studies showed 5% O-18 incorporation into benzaldehyde. Furthermore, C–H oxidation of PhCH2OH and PhCD2OH (∼95%, D enriched) provided a kinetic isotope effect value of 11 which suggests that the initial hydrogen atom abstraction is the rate determining step.3b,8,16 Cyclobutanol as the mechanistic probe provided cyclobutanone exclusively (2e− oxidation product) (the ring opening product 4-hydroxybutanal was not detected) without any O-18 labeling (Scheme 2). These observations suggest that following HAA, an electron transfer mechanism is operational during C–H oxidation of benzyl alcohol.16,17 Subsequently, we studied the C–H oxidation chemistry of 2 using ethylbenzene (Scheme 3), cumene and cyclohexane.8 Cyclohexane produced cyclohexanol (∼15% yield, 52% O-18 enriched) whereas ethyl benzene gave 1-phenyl ethanol (yield, 22%; 60% O-18 labeled).
 | ||
| Scheme 2 Alcohol oxidations by [(N4Py)OMe,MeFeIV(O)]2+ (2). | ||
 | ||
| Scheme 3 Intermediates during reaction of 2 and ethylbenzene. | ||
The ESI-MS data obtained upon addition of ethylbenzene to 2 suggested the formation of iron(III)–hydroxide (3, m/z, 705.150), 1-phenylethanol bound intermediate, [(N4Py)OMe,MeFeII (HO(Me)CHPh)](OTf)+ (4) (m/z, 810.22; Fig. 2g) and iron(III)–alkoxide, [(N4Py)OMe,MeFeIII(O(Me) CHPh)](OTf)+ (5, m/z, 809.215; Fig. 2b and e) (Scheme 3). Most interestingly, the 1-phenylethanol bound intermediate [(N4Py)OMe,MeFeII(HO(Me)CHPh)](OTf)+ (4) formed as a consequence of the radical rebound step was rapidly oxidized by 2 to produce 3 and 5.7 The formation of 5 occurred via the comproportionation reaction of 1 and 2 in the presence of 1-phenyl ethanol. This was further verified by adding 1-phenyl ethanol to a solution of 2 in acetonitrile where both 3 and 5 were simultaneously detected.
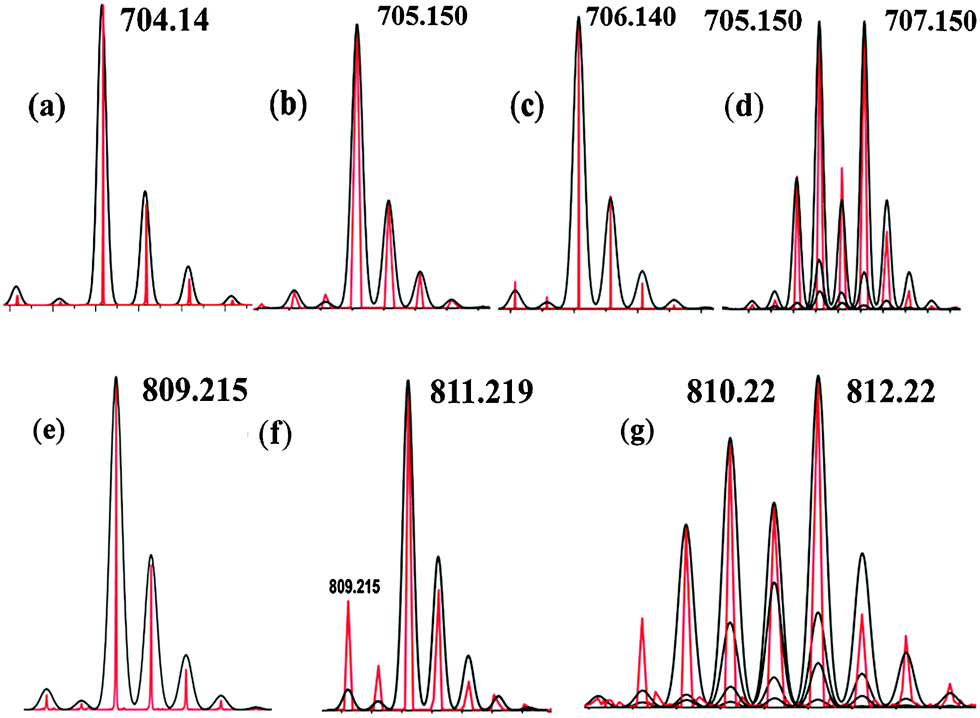 | ||
| Fig. 2 ESI-MS of the intermediates during reaction of 2 and ethylbenzene (red line, experimental and black line, simulated, spectra were recorded after 5 min of addition). ESI-MS of 2 (a), 3 (b), 18-O-2 (c), 18-O-3 (d), 5 (e), 18-O-5 (f), 4 and 18-O-4 (g).8 | ||
Furthermore, the formation of iron(III) complex was confirmed from rhombic signals at g1 = 1.94, g2 = 2.11, g3 = 2.10 and axial signals at g1 = 4.17, g2 = 5.98 by the EPR experiment of a solution of 2 and ethyl benzene (or cumene) (Fig. 3a and b).5,15,18 Replacing ethyl benzene by cumene also showed the formation of iron(III)–hydroxide (3) and iron(III)–alkoxide species, [(N4Py)OMe,MeFeIII(O(Me)2CPh)] (OTf)+ (5a, m/z, 823.21; Scheme 4), which upon 18O labeling shifted by two mass units (m/z, 825.21; Fig. 4a and b) along with 70% O-18 enriched cumyl alcohol.
 | ||
| Fig. 3 EPR spectra (acetonitrile, 77 K) obtained from reaction between 2 and (a) ethylbenzene (b) cumene. | ||
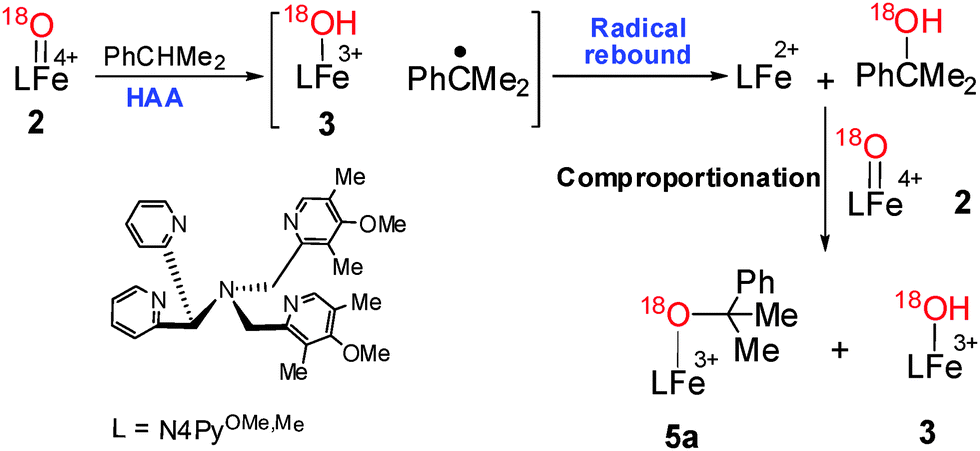 | ||
| Scheme 4 Radical rebound pathway for C–H oxidation of cumene. | ||
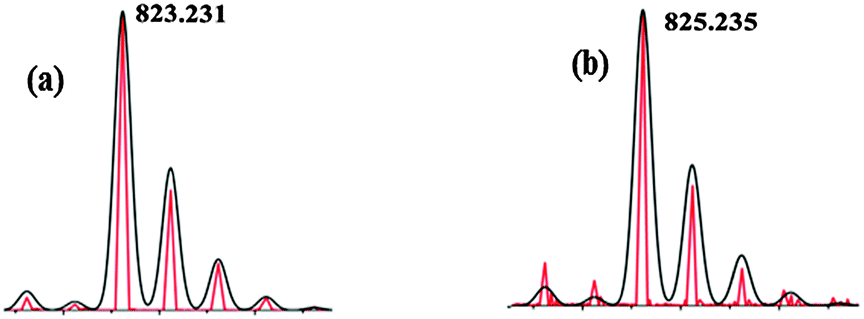 | ||
| Fig. 4 ESI-MS of the intermediates, 5a (a) and 18-O labeled 5a (b) during reaction of 2 and cumene (red line, experimental and black line, simulated). | ||
The formation of 5a was presumed to occur via the comproportionation reaction. This was verified when 2-phenyl-2-propanol was added to a solution of 2; trace amounts of 3 and 5a were observed after 30 min. Notably, a significant amount of these compounds was formed after 16 h. Complex 2 decayed with time to form 1, which in the presence of 2-phenyl-2-propanol and 2 underwent the comproportionation reaction to form 3 and 5a. Expectedly, formation of 3 and 5a (Scheme 4) was observed within 10 minutes via the comproportionation reaction when 2-phenyl-2-propanol was added to a mixture of 1 and 2.
In the presence of ethylbenzene (or cumene), the absorbance vs. time plot for complex 2 (decay profile at 692 nm) was fitted with the pseudo-first order reaction profile (rate constant, k1; Fig. 5).3b,4a,5 A straight line was obtained by plotting the different values of k1 against concentration of the substrate. The slope of this plot yielded the second order rate constant (k2, Fig. 5).8 During the C–H oxidation reaction by 2, cumene reacted slightly faster (k2 ∼ 0.01 M−1 s−1) compared to ethylbenzene (k2 ∼ 0.0021 M−1 s−1) due to the higher strength of the benzylic C–H bond.19 The reaction of cumene with complex 2 occurred at a slightly faster rate (∼5 times, k2 ∼ 0.01 M−1 s−1vs. k2 = 0.002 M−1 s−1 for Fe–N4Py–oxo)4a compared to that for the unsubstituted Fe–N4Py–(oxo) complex. However, the reaction rate of 2 with ethylbenzene is similar to that of its unsubstituted analogue (0.0021 M−1 s−1vs. 0.0031 M−1 s−1 or 0.008 M−1 s−1).4a,5 Notably, during the C–H oxidation reactions of 2 with ethylbenzene, cumene, triphenyl methane, benzyl alcohol and cyclobutanol (500 equiv.) iron(II) were regenerated. Initially, after 1–2 hours of the reaction, 40–60% of iron(II) species was regenerated. After 48 hours of the reaction, iron(II) was obtained quantitatively (∼95%). On the contrary, the unsubstituted [FeII(N4Py)(O)]2+ complex generated ∼95% of the iron(III) species via a dissociative pathway after completion of the reaction with ethyl benzene, cumene and triphenyl methane.5
 | ||
| Fig. 5 (a) UV-vis change at 692 nm for 2, in presence of cumene, (b) kinetics plot for cumene and ethylbenzene. | ||
Cyclohexane oxidation by 2 produced a major amount of iron(III) (∼90%) and minor amount of iron(II) species (10%). Moreover, the cyclohexyl radical was trapped in the form of cyclohexyl bromide upon addition of CCl3Br (or CBr4) during cyclohexane oxidation by 2.8,11 These experimental pieces of evidence suggested that cyclohexane oxidation was likely following a dissociative pathway.
No radical trapped or brominated product was found during the reaction of 2 with ethylbenzene or cumene.11,20 Therefore, the substrate based organic radicals failed to escape from the solvent cage for ethylbenzene and cumene.20c Although radicals formed via the dissociative pathway have been trapped as per the prescient knowledge reported in the literature for non-heme iron(IV)–oxo, our experimental observations suggested that following HAA, the iron(III)–hydroxide and the exogenous substrate based radical may not undergo dissociation (e.g. in the case of ethylbenzene and cumene).21 The reaction followed a radical rebound pathway and produced an iron(II)–alcohol coordinated product that was subsequently oxidized by 2.20c
In summary, we have synthesized an electron rich, room temperature stable and reactive non-heme iron(IV)–oxo species [(N4Py)OMe,MeFeIV(O)](OTf)+ (2). The iron(IV)–oxo derived intermediates like iron(III)–hydroxide (3), iron(III)–alkoxide (5) and substrate-bound iron(II) species (4) were detected from the reaction mixture. The mechanistic switch during C–H oxidation by non-heme iron(IV)–oxo complex 2 mainly depends on the stability of the radical generated after HAA. More stable radicals preferred the electron transfer pathway (Scheme 2), whereas moderately stable radicals underwent the radical rebound pathway (Schemes 3 and 4). The least stable radicals of all (e.g. in case of cyclohexane) underwent the dissociative pathway.
This study was supported by SERB, India (EMR/2015/000164). Financial support received from CSIR-India (fellowships to S. R.) is gratefully acknowledged. DM sincerely thanks Dr. Sayam Sen Gupta (NCL Pune), Dr. Tapan K. Paine (IACS Kolkata) and their group members for stimulating scientific discussions and providing constant support to conduct a number of experiments in their laboratories.
Notes and references
- (a) L. Que, Jr. and W. B. Tolman, Nature, 2008, 455, 333 CrossRef PubMed; (b) M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr., Chem. Rev., 2004, 104, 939 CrossRef CAS PubMed; (c) K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942 CrossRef CAS PubMed; (d) B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947 CrossRef CAS PubMed; (e) J. Rittle and M. T. Green, Science, 2010, 330, 933 CrossRef CAS PubMed; (f) C. Krebs, D. Galonić Fujimori, C. T. Walsh and J. M. Bollinger, Acc. Chem. Res., 2007, 40, 484 CrossRef CAS PubMed; (g) S. Fukuzumi, Coord. Chem. Rev., 2013, 257, 1564 CrossRef CAS PubMed; (h) O. Y. Lyakin and A. A. Shteinman, Kinet. Catal., 2012, 53, 694 CrossRef CAS; (i) E. I. Solomon, S. D. Wong, L. V. Liu, A. Decker and M. S. Chow, Curr. Opin. Chem. Biol., 2009, 13, 99 CrossRef CAS PubMed; (j) X. Shan and L. Que, Jr., J. Inorg. Biochem., 2006, 100, 421 CrossRef CAS PubMed; (k) A. R. McDonald and L. Que Jr, Coord. Chem. Rev., 2013, 257, 414 CrossRef CAS PubMed.
- (a) M. Girhard and V. B. Urlacher, Modern Oxidation Methods, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, p. 421 Search PubMed; (b) J. T. Groves, J. Inorg. Biochem., 2006, 100, 434 CrossRef CAS PubMed; (c) D. Hamdane, H. Zhang and P. Hollenberg, Photosynth. Res., 2008, 98, 657 CrossRef CAS PubMed; (d) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932 CrossRef CAS PubMed; (e) V. B. Urlacher and M. Girhard, Trends Biotechnol., 2012, 30, 26 CrossRef CAS PubMed.
- (a) A. N. Biswas, M. Puri, K. K. Meier, W. N. Oloo, G. T. Rohde, E. L. Bominaar, E. Münck and L. Que, Jr., J. Am. Chem. Soc., 2015, 137, 2428 CrossRef CAS PubMed; (b) M. Ghosh, K. K. Singh, C. Panda, A. Weitz, M. P. Hendrich, T. J. Collins, B. B. Dhar and S. Sen Gupta, J. Am. Chem. Soc., 2014, 136, 9524 CrossRef CAS PubMed; (c) K. K. Singh, M. K. Tiwari, M. Ghosh, C. Panda, A. Weitz, M. P. Hendrich, B. B. Dhar, K. Vanka and S. Sen Gupta, Inorg. Chem., 2015, 54, 1535 CrossRef CAS PubMed; (d) P. Barman, A. K. Vardhaman, B. Martin, S. J. Wörner, C. V. Sastri and P. Comba, Angew. Chem., Int. Ed., 2015, 54, 2095 CrossRef CAS PubMed; (e) T. A. Jackson, J.-U. Rohde, M. S. Seo, C. V. Sastri, R. DeHont, A. Stubna, T. Ohta, T. Kitagawa, E. Münck, W. Nam and L. Que, Jr., J. Am. Chem. Soc., 2008, 130, 12394 CrossRef CAS PubMed; (f) K. Ray, J. England, A. T. Fiedler, M. Martinho, E. Münck and L. Que, Jr., Angew. Chem., Int. Ed., 2008, 47, 8068 CrossRef CAS PubMed; (g) S. A. Wilson, J. Chen, S. Hong, Y.-M. Lee, M. Clémancey, R. Garcia-Serres, T. Nomura, T. Ogura, J.-M. Latour, B. Hedman, K. O. Hodgson, W. Nam and E. I. Solomon, J. Am. Chem. Soc., 2012, 134, 11791 CrossRef CAS PubMed; (h) A. R. McDonald, Y. Guo, V. V. Vu, E. L. Bominaar, E. Munck and L. Que, Jr., Chem. Sci., 2012, 3, 1680 RSC; (i) F. T. de Oliveira, A. Chanda, D. Banerjee, X. Shan, S. Mondal, L. Que Jr., E. L. Bominaar, E. Münck and T. J. Collins, Science, 2007, 315, 835 CrossRef PubMed; (j) S. Kundu, J. V. K. Thompson, A. D. Ryabov and T. J. Collins, J. Am. Chem. Soc., 2011, 133, 18546 CrossRef CAS PubMed.
- (a) J. Kaizer, E. J. Klinker, N. Y. Oh, J.-U. Rohde, W. J. Song, A. Stubna, J. Kim, E. Münck, W. Nam and L. Que, Jr., J. Am. Chem. Soc., 2003, 126, 472 CrossRef PubMed; (b) C. V. Sastri, S. M. Sook, P. M. Joo, K. K. Mook and W. Nam, Chem. Commun., 2005, 1405 RSC; (c) H. Hirao, D. Kumar, L. Que, Jr. and S. Shaik, J. Am. Chem. Soc., 2006, 128, 8590 CrossRef CAS PubMed; (d) D. Kumar, S. P. de Visser and S. Shaik, J. Am. Chem. Soc., 2003, 125, 13024 CrossRef CAS PubMed; (e) D. Kumar, H. Hirao, L. Que, Jr. and S. Shaik, J. Am. Chem. Soc., 2005, 127, 8026 CrossRef CAS PubMed; (f) D. Kumar, B. Karamzadeh, G. N. Sastry and S. P. de Visser, J. Am. Chem. Soc., 2010, 132, 7656 CrossRef CAS PubMed; (g) W. Nam, Acc. Chem. Res., 2007, 40, 522 CrossRef CAS PubMed; (h) W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146 CrossRef CAS PubMed; (i) S. Ye and F. Neese, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1228 CrossRef CAS PubMed; (j) Y. J. Jeong, Y. Kang, A.-R. Han, Y.-M. Lee, H. Kotani, S. Fukuzumi and W. Nam, Angew. Chem., Int. Ed., 2008, 47, 7321 CrossRef CAS PubMed; (k) W. Lai, C. Li, H. Chen and S. Shaik, Angew. Chem., Int. Ed., 2012, 51, 5556 CrossRef CAS PubMed; (l) C. Geng, S. Ye and F. Neese, Angew. Chem., Int. Ed., 2010, 49, 5717 CrossRef CAS PubMed; (m) P. Comba, M. Maurer and P. Vadivelu, Inorg. Chem., 2009, 48, 10389 CrossRef CAS PubMed.
- K.-B. Cho, X. Wu, Y.-M. Lee, Y. H. Kwon, S. Shaik and W. Nam, J. Am. Chem. Soc., 2012, 134, 20222 CrossRef CAS PubMed.
- (a) X. Wu, M. S. Seo, K. M. Davis, Y.-M. Lee, J. Chen, K.-B. Cho, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2011, 133, 20088 CrossRef CAS PubMed; (b) E. Kwon, K.-B. Cho, S. Hong and W. Nam, Chem. Commun., 2014, 50, 5572 RSC.
- T. Kojima, K. Nakayama, K. Ikemura, T. Ogura and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11692 CrossRef CAS PubMed.
- See ESI†.
- M. Lubben, A. Meetsma, E. C. Wilkinson, B. L. Feringa and L. Que Jr, Angew. Chem., Int. Ed., 1995, 34, 1512 CrossRef CAS PubMed.
- E. J. Klinker, J. Kaizer, W. W. Brennessel, N. L. Woodrum, C. J. Cramer and L. Que Jr, Angew. Chem., Int. Ed., 2005, 44, 3690 CrossRef CAS PubMed.
- G. Roelfes, M. Lubben, K. Chen, R. Y. N. Ho, A. Meetsma, S. Genseberger, R. M. Hermant, R. Hage, S. K. Mandal, V. G. Young, Y. Zang, H. Kooijman, A. L. Spek, L. Que Jr and B. L. Feringa, Inorg. Chem., 1999, 38, 1929 CrossRef CAS PubMed.
- (a) G. Roelfes, V. Vrajmasu, K. Chen, R. Y. N. Ho, J.-U. Rohde, C. Zondervan, R. M. la Crois, E. P. Schudde, M. Lutz, A. L. Spek, R. Hage, B. L. Feringa, E. Münck and L. J. Que Jr, Inorg. Chem., 2003, 42, 2639 CrossRef CAS PubMed; (b) D. Wang, K. Ray, M. J. Collins, E. R. Farquhar, J. R. Frisch, L. Gomez, T. A. Jackson, M. Kerscher, A. Waleska, P. Comba, M. Costas and L. Que, Jr, Chem. Sci., 2013, 4, 282 RSC; (c) M. J. Collins, K. Ray and L. J. Que Jr, Inorg. Chem., 2006, 45, 8009 CrossRef CAS PubMed.
- (a) A. Decker, J.-U. Rohde, E. J. Klinker, S. D. Wong, L. Que Jr and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 15983 CrossRef CAS PubMed; (b) A. Decker, J.-U. Rohde, L. Que Jr and E. I. Solomon, J. Am. Chem. Soc., 2004, 126, 5378 CrossRef CAS PubMed.
- M. S. Seo, J.-H. In, S. O. Kim, N. Y. Oh, J. Hong, J. Kim, L. Que Jr and W. Nam, Angew. Chem., Int. Ed., 2004, 43, 2417 CrossRef CAS PubMed.
- J. J. Braymer, K. P. O'Neill, J.-U. Rohde and M. H. Lim, Angew. Chem., Int. Ed., 2012, 51, 5376 CrossRef CAS PubMed.
- N. Y. Oh, Y. Suh, M. J. Park, M. S. Seo, J. Kim and W. Nam, Angew. Chem., Int. Ed., 2005, 44, 4235 CrossRef CAS PubMed.
- S. Kumar, A. S. Faponle, P. Barman, A. K. Vardhaman, C. V. Sastri, D. Kumar and S. P. de Visser, J. Am. Chem. Soc., 2014, 136, 17102 CrossRef CAS PubMed.
- (a) Y. Nishida, Y. Morimoto, Y.-M. Lee, W. Nam and S. Fukuzumi, Inorg. Chem., 2013, 52, 3094 CrossRef CAS PubMed; (b) A. Draksharapu, D. Angelone, M. G. Quesne, S. K. Padamati, L. Gómez, R. Hage, M. Costas, W. R. Browne and S. P. de Visser, Angew. Chem., Int. Ed., 2015, 54, 4357–4361 CrossRef CAS PubMed; (c) J. Yoon, S. A. Wilson, Y. K. Jang, M. S. Seo, K. Nehru, B. Hedman, K. O. Hodgson, E. Bill, E. I. Solomon and W. Nam, Angew. Chem., Int. Ed., 2009, 48, 1257 CrossRef CAS PubMed; (d) S. Rana, S. Bag, T. Patra and D. Maiti, Adv. Synth. Catal., 2014, 356, 2453 CrossRef CAS PubMed.
- J. R. Bryant and J. M. Mayer, J. Am. Chem. Soc., 2003, 125, 10351 CrossRef CAS PubMed.
- (a) T. Kojima, R. A. Leising, S. Yan and L. Que Jr, J. Am. Chem. Soc., 1993, 115, 11328 CrossRef CAS; (b) L. Mathew and J. Warkentin, Can. J. Chem., 1988, 66, 11 CrossRef CAS; (c) J. T. Groves and T. E. Nemo, J. Am. Chem. Soc., 1983, 105, 6243 CrossRef CAS.
- A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1051845. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04803f |
| This journal is © The Royal Society of Chemistry 2015 |