
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis of the heterocubane cluster [{CpMn}4(μ3-P)4] as a tetrahedral shaped starting material for the formation of polymeric coordination compounds†‡§
Sebastian
Heinl
a,
Konrad
Kiefer
b,
Gábor
Balázs
a,
Claudia
Wickleder
b and
Manfred
Scheer
*a
aUniversity of Regensburg, Universitätsstr. 31, 93040 Regensburg, Germany. E-mail: manfred.scheer@chemie.uni-regensburg.de; Fax: +49 941 943 4439; Tel: +49 941 943 4440
bAnorganische Chemie II, Naturwissenschaftlich-Technische Fakultät, Department Chemie-Biologie, Universität Siegen, Adolf-Reichwein-Straße, D-57068 Siegen, Germany. E-mail: wickleder@chemie.uni-siegen.de; Fax: +49 271 4702555
First published on 17th July 2015
Abstract
Thermolysis of [CpMn(η6-cht)] with P4 in 1,3-diisopropylbenzene leads to the formation of the heterocubane [Cp4Mn4P4] (1) in high yields, as a rare example of ‘naked’ phosphorus containing complexes of manganese. Compound 1 is characterized and studied by DFT calculations and reflection measurements. 1D coordination polymers [{(CpMn)4(μ3-P)4}(CuX)]n (2-Cl: X = Cl; 2-Br: X = Br) are obtained in the reaction with CuX. Furthermore, it is shown that all four P atoms in 1 can be addressed for a coordination towards cymantrene resulting in [{(CpMn)4(μ3-P)4}{CpMn(CO)2}n] (3a: n = 1; 3b: n = 2; 3c: n = 3; 3d: n = 4), and shows that 1 is a tetra-topic building block in coordination chemistry.
In the last few decades, the field of Pn ligand complexes1 has been well established in chemistry.2 Within this area, we demonstrated that Pn ligand complexes in combination with monovalent coinage metal salts are perfect starting materials to construct supramolecular assemblies. Especially, using the complexes [{CpMo(CO)2}2(μ,η2:2-P2)] and [Cp*Fe(η5-P5)] (Cp* = C5Me5) together with CuX (X = Cl, Br, I), a wide spectrum of pseudo-0D,3 1D4 or 2D4a,d frameworks could be realized. For the formation of 3D architectures, ideally multitopic linkers with an appropriate symmetry (e.g. Td, Oh) would be needed. All attempts to construct 3D scaffolds by using Pn ligand complexes have failed so far.5
The existence of a restricted number of clusters and complexes of manganese with substituent-free P atoms motivated us to prepare such compounds.6 Very recently we reported on the synthesis of the two triple decker sandwich complexes [{CpBIGMn}2(μ,η5:5-P5)] and [{CpBIGMn}2(μ,η2:2-P2)2] (CpBIG = C5(4-nBuC6H4)5) via the thermolysis of [CpBIGMn(cht)] (cht = cycloheptatriene) with P4.6a Substitution of the sterically demanding CpBIG ligand by the usual Cp ligand should alter the product outcome dramatically.
In general, Mn4+ ions are very promising candidates for future warm white LEDs due to their emission in the red range of the visible spectrum and large intensities in some host lattices, but their luminescence is mainly investigated by doping in oxidic or fluoridic solid state materials.7 For the cluster presented in this communication the electronic situation is quite different, but the formal manganese oxidation state is also IV. Therefore we investigated the optical properties of 1.
Herein we report on the synthesis and characterization of the heterocubane cluster [(CpMn)4(μ3-P)4] (1) and the first coordination studies of 1 with CuX (X = Cl, Br) resulting in 1D coordination polymers. It is also shown that the coordination of all four P atoms in 1 towards metals can be generally achieved.
The co-thermolysis (20 min) of [CpMn(η6-cht)] (cht = cycloheptatriene) and P4 in 1,3-diisopropylbenzene (DIB) results in the formation of [Cp4Mn4P4] (1) (eqn (1)). After solvent removal, extraction with CH2Cl2 and cooling to −35 °C, 1 is obtained as dark red to brown needle shaped crystals in yields of about 73%. Decomposition is observed at longer reaction times (metal mirror).
 | (1) |
Both the 1H NMR and the 13C{1H} NMR spectra (CD2Cl2) of 1 show singlets at 4.58 ppm and 82.7 ppm, respectively, in the expected regions. In contrast, the 31P{1H} NMR spectrum of 1 shows an extremely low-field shifted singlet at 1079.8 ppm (slightly broadened, ω1/2 = 31.3 Hz). Astonishingly, the signal is about 230 ppm to 600 ppm low field shifted compared to A–D.
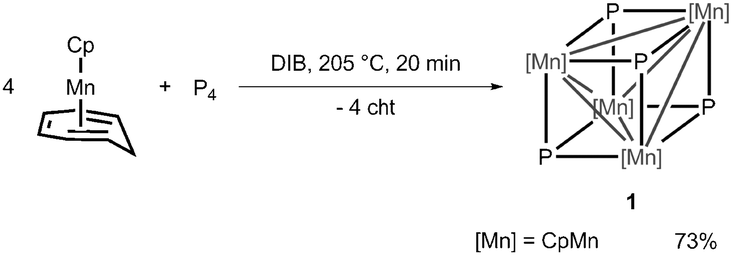
The NMR data indicate a highly symmetric molecule, which is confirmed by single crystal X-ray diffraction (Fig. 1). Compound 1 crystallizes with one solvent molecule CH2Cl2 from concentrated CH2Cl2 solutions in the monoclinic space group C2/c. The core of 1 can be described as a Mn4 tetrahedron, wherein the faces are capped by P atoms, resulting in a heterocubane structure. In comparison, the central cores of the structures of A, C and D are much more distorted from a perfect cube structure than 1 is. The four Mn atoms in 1 are coordinatively saturated by Cp ligands. The Mn–Mn and Mn–P bond lengths vary from 2.6914(8) Å to 2.7320(6) Å and from 2.2249(9) Å to 2.2411(10) Å, respectively.
 | ||
| Fig. 1 Molecular structure of 1 in the crystal. For clarity only one molecule of the asymmetric unit is depicted, C atoms are shown in ‘wireframe’ model and H atoms and solvent molecules are omitted. Ellipsoids are drawn at the 50% probability level. | ||
The number of valence electrons of the Mn atoms can be calculated classically to be 18 and the Mn centers have a formal oxidation state of +4, but 1 could be better described to contain a [Mn4P4]4+ cluster. To gain more insight into the electronic structure of 1 DFT calculations have been performed (Fig. 2). The geometry of 1 has been optimized for different spin states. According to the calculations 1 has a singlet ground state (Table S5, ESI§). The triplet spin state is with 70.3 kJ mol−1 higher in energy. All other spin states are more than 100 kJ mol−1 higher in energy. Interestingly, the spin state of 1 has a strong influence on the geometry. In the singlet spin state the optimized geometry is very close to the observed experimental one. E.g. it possesses short Mn–Mn distances (2.705 Å) which are very similar to the experimentally determined distance of 2.713 Å (average). By stepwise increasing the spin multiplicity the elongation of one Mn–Mn distance is observed. For example in the triplet spin state there is one longer Mn–Mn distance of 3.189 Å and five shorter ones of 2.714 Å (average). In comparison with the experimentally found parameters it is indicative that 1 has a singlet spin state. The Wiberg bond index indicates a Mn–Mn bond order of 0.33, while the Mn–P bond order is close to unity (0.92). The molecular orbitals (MO) as well as the localized molecular orbitals (LMO) of 1 show the presence of Mn2P2 four centre bonds (2e−) and the presence of lone pairs at the phosphorus atoms (Fig. 2 and Fig. S18 and S19, ESI§).
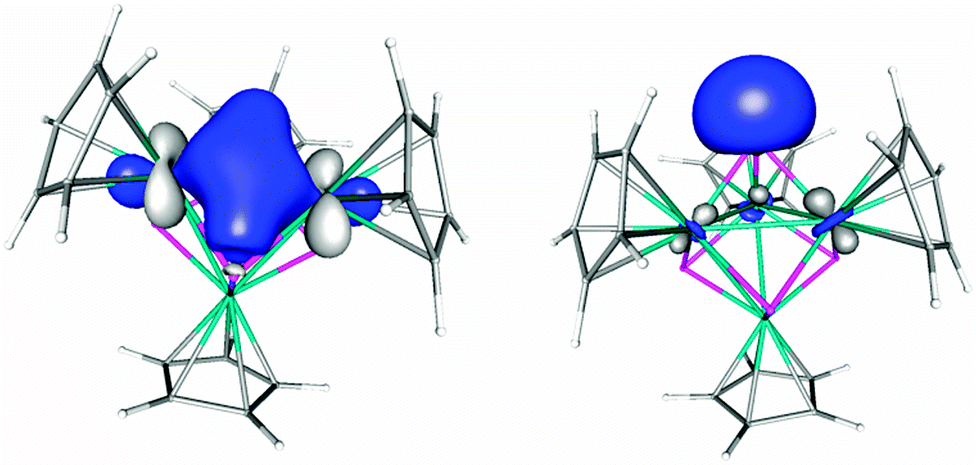 | ||
| Fig. 2 Selected localized molecular orbitals representing the Mn–P bonding (left) and the lone pair of the phosphorus atoms (right) in [(CpMn)4(P)4] (1) in singlet spin state. Calculated at the BP86/def2-TZVP level of theory. | ||
The results of the optical measurements fit well to the findings of the theoretical calculations. No emission of compound 1 could be observed neither in the visible range nor in the IR range (down to 1600 nm) even at 10 K. This can be explained by the different spin multiplicities of the ground and excited states which causes low emission intensity due to the spin selection rule. On the other hand the large number of electronic states in relatively small energetic distances causes non-radiative transitions rather than emission of radiation.
The room temperature reflection spectrum of 1 is depicted in Fig. 3. Besides some vibrational transition in the IR range the electronic transitions of the basis of the DFT calculations described above can be detected in the reflection spectrum. In detail, the first singlet–triplet electronic transition at about 1700 nm and also the one of the next higher electronic state identified by the DFT calculations to be at about 930 nm could be detected (Fig. 3).
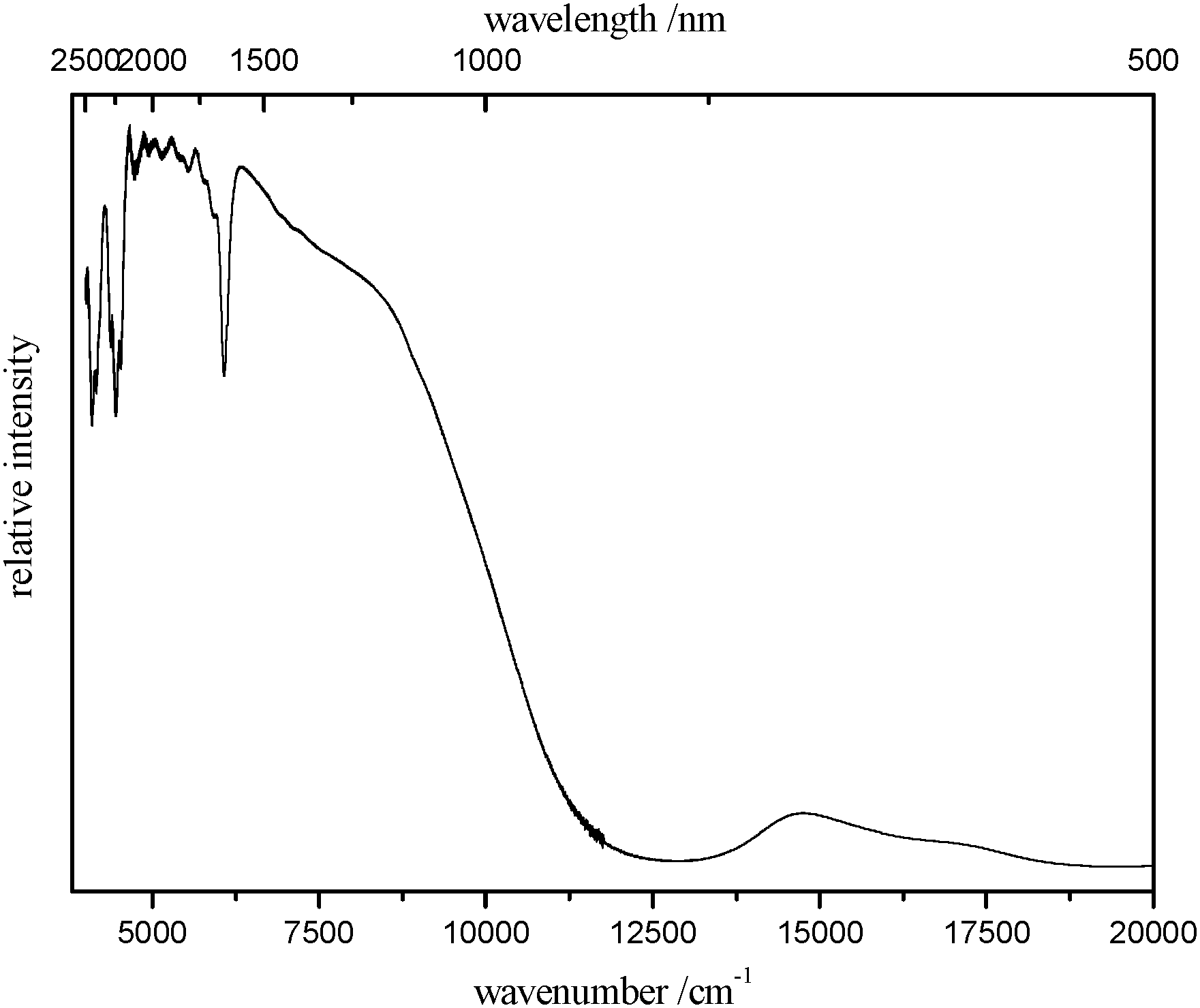 | ||
Fig. 3 Room temperature reflection spectrum of 1. Cf. calculated electronic transitions: 1702 nm (5874 cm−1) for singlet–triplet and 935 nm (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 693 cm−1) for singlet–quintet (BP86/def2-TZVP level of theory). 693 cm−1) for singlet–quintet (BP86/def2-TZVP level of theory). | ||
The four P atoms in 1 are pyramidal and bear a lone pair each. This should be a perfect building block for the formation of three-dimensional networks. By slow diffusion of CH3CN solutions of CuX (X = Cl, Br) into CH2Cl2 solutions of 1, some black needle-shaped crystals and plenty of off-white to brown voluminous, amorphous powder were obtained. The reaction with a 1:1 stoichiometry of 1 and CuX results in a still coloured mother liquor solution and a larger amount of crystals. In contrast, a 1:5 stoichiometry results in a colourless solution and more powder. The crystals as well as the powder are totally insoluble in all common solvents.
The crystals were characterized by single crystal X-ray diffraction. The crystal structures illustrate zigzag like 1D coordination polymers with the sum formulae [{(CpMn)4(μ3-P)4}(CuX)]n (2-Cl: X = Cl; 2-Br: X = Br) (Fig. 4).16 However, in the case of 2-Cl, one molecule CH2Cl2 co-crystallizes per formula unit, which is not the case for 2-Br. Two of the P atoms in 2-Cl and 2-Br coordinate to CuX units resulting in a trigonal planar coordination of the Cu atoms. In 2-Br a disorder of the Br atom over two positions was observed. With angular sums around the Cu centres of 359.8° for 2-Cl and 356.8° and 360.0°, respectively, for 2-Br this is an indication for almost perfect planarity. The polymeric strands are orientated in parallel with respect to each other.
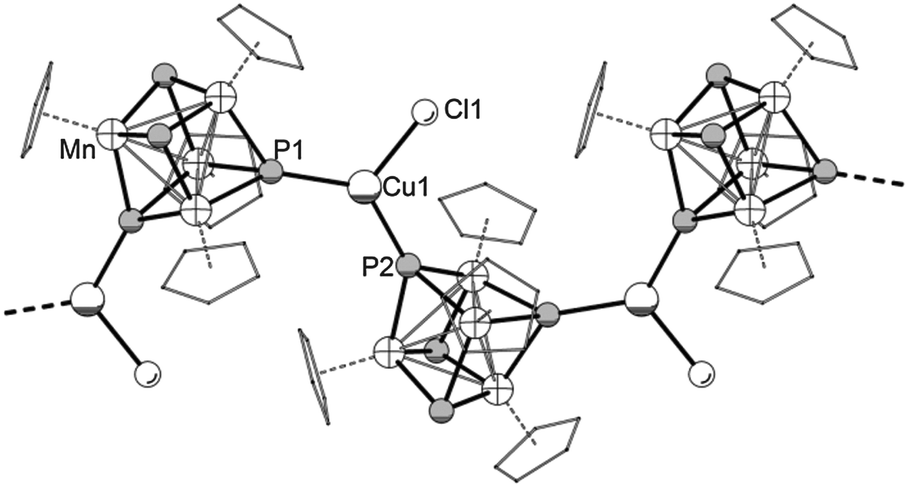 | ||
| Fig. 4 Section of the 1D zigzag chain [{(CpMn)4(μ3-P)4}(CuCl)]n (2-Cl) in the crystal. View along crystallographic c-axis. For clarity reasons the C atoms are shown in ‘wireframe’ model and H atoms and solvent molecules are omitted. Because of the isostructural constitution of 2-Br, its structure is not depicted. Selected bond lengths [Å] and angles [°] of 2-Cl: P1–Cu1 2.248(1), P2–Cu1 2.252(1), Cu1–Cl1 2.245(2), P1–Cu1–P2 129.64(5), P1–Cu1–Cl1 117.26(5), P2–Cu1–Cl1 112.85(5), av. Mn–P 2.228, av. Mn–Mn 2.720. Selected bond lengths [Å] and angles [°] of 2-Br (for labeling see the ESI§): P1–Cu1 2.248(3), P4–Cu1 2.252(3), Cu1–Br1 2.372(8), Cu1–Br2 2.324(5), P1–Cu1–P4 129.0(1), P1–Cu1–Br1 119.8(2), P1–Cu1–Br2 115.1(1), P4–Cu1–Br1 108.0(2), P4–Cu1–Br2 115.9(1), av. Mn–P 2.231, av. Mn–Mn 2.718. | ||
The off-white powder was found to be amorphous, as indicated by X-ray powder diffraction. Also 31P{1H} MAS NMR did not show any signals. To further analyse the amorphous powder, it was washed several times with CH3CN and dried carefully in a vacuum. To get a hint of the composition, elemental analyses were carried out. With the assumption of complete removal of the solvent and excess of CuX, the result fits best for a 1:CuX ratio of roughly 1:3. This might be a hint for the coordination of more than two of P atoms to CuX and the formation of a 3D structure.
For the formation of three-dimensional networks, the coordination of all four P atoms in 1 is necessary. To check whether this is possible at all, 1 was reacted with [CpMn(CO)2(thf)], and in fact, the coordination of up to four P atoms can be achieved, resulting in the formation of [{(CpMn)4(μ3-P)4}{CpMn(CO)2}n] (3a: n = 1; 3b: n = 2; 3c: n = 3; 3d: n = 4). In a 1:1 stoichiometry the mono- and di-coordination product is obtained and in a 1:4 stoichiometry the tri- and tetra-coordination derivative occurs. Compounds 3a–d can be separated by column chromatography. However, complex 3a elutes together with the unreacted cluster 1.
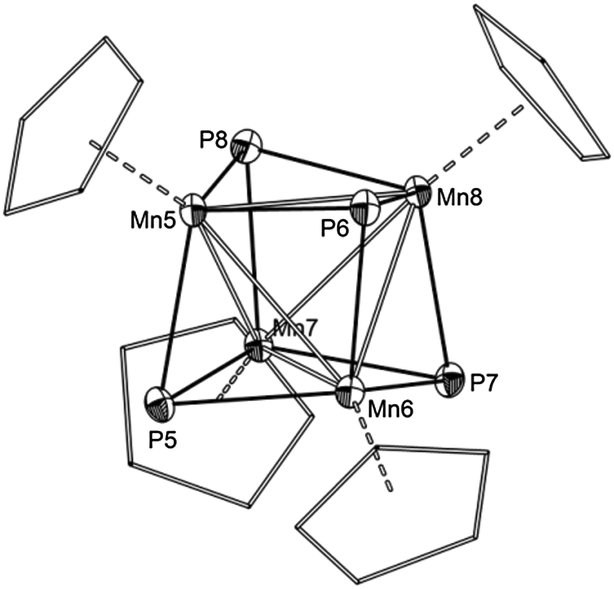
All four compounds were crystallized as solvates from concentrated CH2Cl2 solutions. The molecular structures are depicted in Fig. 5. The average bond lengths between the P atoms and the {CpMn(CO)2} fragments increase with the degree of coordination (3a: 2.260 Å; 3b: 2.262 Å; 3c: 2.272 Å; 3d: 2.287 Å). The steric parameters of the heterocubane cores are also affected by the coordination. While the average Mn–Mn and Mn–P bond lengths in 3a are almost identical in comparison to 1, they increase steadily from 3b to 3d (1: av. Mn–P 2.235 Å, av. Mn–Mn 2.713 Å; 3d: av. Mn–P 2.258 Å, av. Mn–Mn 2.757 Å). The increase in the Mn–Mn and Mn–P bond lengths effectively results in an expansion of the heterocubane cages. The volumes of the Mn4P4 cages vary in the same way as the bond lengths. With just one coordination it remains almost unaffected, but starting from the second coordination in 3b the volume increases constantly (V(1) = 9.159 Å3; V(3a) = 9.151 Å3; V(3b) = 9.333 Å3; V(3c) = 9.372 Å3; V(3d) = 9.490 Å3).17 The core overall expands by 3.5%. This observation might be explained by the electron withdrawing effect of the {CpMn(CO)2} fragments.
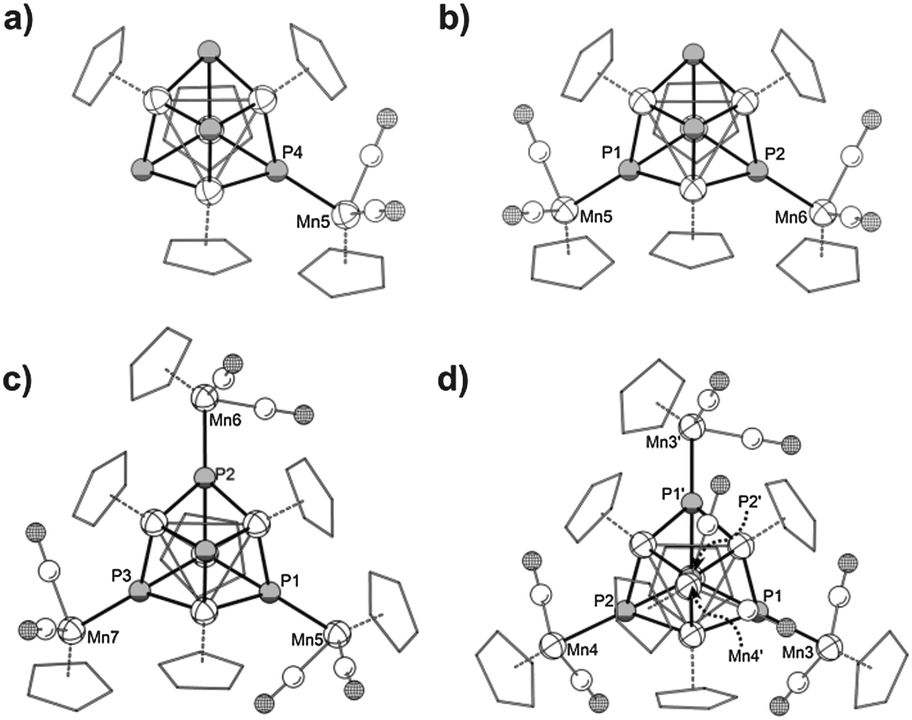 | ||
| Fig. 5 Molecular structures in the crystals: (a) 3a, (b) 3b, (c) 3c, (d) 3d. For clarity the Cp carbon atoms are shown in ‘wireframe’ model and H atoms and solvent molecules are omitted. In the case of the disordered molecule 3b only the main part is depicted. Selected Mn–P bond lengths [Å] in 3a: Mn5–P4 2.2603(8), av. Mn–P 2.234, av. Mn–Mn 2.719; in 3b: Mn5–P1 2.2576(10), Mn6–P2 2.2672(8), av. Mn–P 2.246, av. Mn–Mn 2.740; in 3c: Mn5–P1 2.2779(8), Mn6–P2 2.2778(7), Mn7–P3 2.2616(9) av. Mn–P 2.245, av. Mn–Mn 2.748; in 3d: Mn3–P1 2.2928(7), Mn4–P2 2.2818(11), av. Mn–P 2.258, av. Mn–Mn 2.757. Average values are calculated only from the heterocubane cores. | ||
In conclusion, we have reported on the synthesis of the novel cluster [Cp4Mn4P4] (1) with a central heterocubane structure in good yields, which is a good building block for promoting higher-dimensional aggregation. Compound 1 was characterized by X-ray diffraction, spectroscopic measurements and DFT calculations. Reaction with CuX (X = Cl, Br) leads to the formation of 1D coordination polymers with the zigzag-like structural motif. As a proof of concept it was demonstrated that all four P atoms in 1 can be addressed for coordination, which was exemplified by the coordination of {CpMn(CO)2} moieties. An interesting cluster volume expansion is observed, correlating with the degree of coordination. The formation of 3D networks starting from 1 will be the focus of future work.
The European Research Council (ERC) is acknowledged for the support in the SELFPHOS project AdG-2013-330072. The authors are grateful to COST action CM1302 for general support. S. Heinl is grateful to the Fonds der Chemischen Industrie for a PhD fellowship.
Notes and references
- Complexes, mainly of transition metals, with unsubstituted Pn ligands. P atoms are only bound to the metal atoms and/or other P atoms.
- (a) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed; (b) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed; (c) M. Scheer, G. Balazs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed; (d) N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342–1359 CrossRef CAS.
- (a) J. Bai, A. V. Virovets and M. Scheer, Science, 2003, 300, 781–783 CrossRef CAS PubMed; (b) M. Scheer, J. Bai, B. P. Johnson, R. Merkle, A. V. Virovets and C. E. Anson, Eur. J. Inorg. Chem., 2005, 4023–4026 CrossRef CAS; (c) M. Scheer, A. Schindler, R. Merkle, B. P. Johnson, M. Linseis, R. Winter, C. E. Anson and A. V. Virovets, J. Am. Chem. Soc., 2007, 129, 13386–13387 CrossRef CAS PubMed.
- (a) J. Bai, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2002, 41, 1737–1740 CrossRef CAS; (b) M. Scheer, L. J. Gregoriades, A. V. Virovets, W. Kunz, R. Neueder and I. Krossing, Angew. Chem., Int. Ed., 2006, 45, 5689–5693 CrossRef CAS PubMed; (c) S. Welsch, L. J. Gregoriades, M. Sierka, M. Zabel, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2007, 46, 9323–9326 CrossRef CAS PubMed; (d) F. Dielmann, A. Schindler, S. Scheuermayer, J. Bai, R. Merkle, M. Zabel, A. V. Virovets, E. V. Peresypkina, G. Brunklaus, H. Eckert and M. Scheer, Chem. – Eur. J., 2012, 18, 1168–1179 CrossRef CAS PubMed; (e) M. Scheer, L. Gregoriades, J. Bai, M. Sierka, G. Brunklaus and H. Eckert, Chem. – Eur. J., 2005, 11, 2163–2169 CrossRef CAS PubMed; (f) B. Attenberger, S. Welsch, M. Zabel, E. Peresypkina and M. Scheer, Angew. Chem., Int. Ed., 2011, 50, 11516–11519 CrossRef CAS PubMed; (g) J. Bai, E. Leiner and M. Scheer, Angew. Chem., Int. Ed., 2002, 41, 783–786 CrossRef CAS.
- Very recently we were able to construct the first 3D scaffolds containing Pn ligand complexes, however by using additional multitopic organic linkers; cf. B. Attenberger, E. V. Peresypkina and M. Scheer, Inorg. Chem., 2015, 54, 7021–7029 CrossRef CAS PubMed.
- Usually, cymantrene fragment coordinated compounds are known, with the exception of: (a) S. Heinl, G. Balázs, M. Bodensteiner and M. Scheer, Dalton Trans., 2015 10.1039/C5DT01750E; (b) A. J. Bridgeman, M. J. Mays and A. D. Woods, Organometallics, 2001, 20, 2932–2935 CrossRef CAS; (c) A. Strube, J. Heuser, G. Huttner and H. Lang, J. Organomet. Chem., 1988, 356, C9–C11 CrossRef CAS; (d) M. Baudler and T. Etzbach, Angew. Chem., Int. Ed., 1991, 30, 580–582 CrossRef.
- (a) W. Lu, W. Lv, Q. Zhao, M. Jiao, B. Shao and H. You, Inorg. Chem., 2014, 53, 11985–11990 CrossRef CAS PubMed; (b) M. Peng, X. Yin, P. A. Tanner, M. G. Brik and P. Li, Chem. Mater., 2015, 27, 2938–2945 CrossRef; (c) L.-L. Wei, C. C. Lin, M.-H. Fang, M. G. Brik, S.-F. Hu, H. Jiao and R.-S. Liu, J. Mater. Chem. C, 2015, 3, 1655–1660 RSC.
- (a) G. L. Simon and L. F. Dahl, J. Am. Chem. Soc., 1973, 95, 2175–2183 CrossRef CAS; (b) O. J. Scherer, S. Weigel and G. Wolmershäuser, Chem. – Eur. J., 1998, 4, 1910–1916 CrossRef CAS.
- M. Scheer, E. Leiner, P. Kramkowski, M. Schiffer and G. Baum, Chem. – Eur. J., 1998, 4, 1917–1923 CrossRef CAS.
- H. Schumann and H. Benda, Angew. Chem., Int. Ed., 1968, 7, 813 CrossRef CAS.
- O. J. Scherer, G. Kemény and G. Wolmershäuser, Chem. Ber., 1995, 128, 1145–1148 CrossRef CAS.
- O. J. Scherer, J. Braun and G. Wolmershäuser, Chem. Ber., 1990, 123, 471–475 CrossRef CAS.
- M. Herberhold, G. Frohmader and W. Milius, J. Organomet. Chem., 1996, 522, 185–196 CrossRef CAS.
- S. Reisinger, M. Bodensteiner, E. M. Pineda, J. J. W. McDouall, M. Scheer and R. A. Layfield, Chem. Sci., 2014, 5, 2443–2448 RSC.
- B. P. Johnson, M. Schiffer and M. Scheer, Organometallics, 2000, 19, 3404–3409 CrossRef CAS.
- The obtained crystals of the reactions with a different stoichiometry show the same crystal structures.
- The heterocubane scaffold is broken up into one Mn4 pyramid and four Mn3P pyramids for the volume calculations (Heron's formula and V = 1/3 × b × h).
Footnotes |
| † Parts of the presented results are already part of the doctoral thesis of S. Heinl, University of Regensburg (Regensburg, Germany), 2014. |
| ‡ Dedicated to Professor Todd Marder on the occasion of his 60th birthday. |
| § Electronic supplementary information (ESI) available: Full crystallographic data, NMR spectra. CCDC 1404879–1404885. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04800a |
| This journal is © The Royal Society of Chemistry 2015 |