
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A neutral low-coordinate heterocyclic bismuth-tin species†
C.
Hering-Junghans
a,
A.
Schulz
*ab and
A.
Villinger
a
aInstitut für Chemie, Universität Rostock, Albert-Einstein-Str. 3a, 18059 Rostock, Germany. E-mail: axel.schulz@uni-rostock.de
bAbteilung Materialdesign, Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
First published on 23rd July 2015
Abstract
The reaction of distannadiazane bearing bulky RAr*-groups (RAr* = C6H2{C(H)Ph2}2R-2,6,4; R = iPr, tBu) with ECl3 (E = Sb, Bi) was studied resulting in the isolation of previously unknown N,N-bis(dichloropnictino)amines (3) and a novel heterocyclic carbenoid bismuth species (4) bearing a Bi(III) and a Sn(IV) center. The structure and bonding was investigated by means of X-ray structure elucidations and DFT calculations.
Pnictogen–nitrogen heterocycles of the type [XE(μ-NR)]2 (E = P, As, Sb, Bi; species I in Fig. 1) are valuable starting materials for preparative E–N chemistry.1 Usually, [ClE(μ-NR)]2 (E = P, As) is prepared from RN(ECl2)H in a base-assisted (e.g. NEt3) cyclization,2 however, for the heavier analogs this strategy works poorly. For example, [ClBi(μ-NTer)]2 (Ter = terphenyl = 2,6-bis-(2,4,6-trimethylphenyl)phenyl) was initially obtained in moderate yields of 45% besides large amounts of ClBi(N(H)Ter)2.3 In analogy to Veith's synthesis of [Me2SiE(μ-NtBu)2]+ (II in Fig. 1),4 our group succeeded in establishing a straightforward route towards the synthesis of [ClE(μ-NTer)]2 (E = Sb, Bi), based on the trans-metalation of the respective tin precursor.5 Now highly reactive cyclo-1,3-dipnicta-2,4-diazenium salts of the type [E(ClE)(μ-NTer)2]+ (E = P, As,6 Sb, Bi;5 III in Fig. 1) can be obtained by chloride abstraction from [ClE(μ-NTer)]2 by means of Lewis acids such as GaCl3. A new area of research opened up with the isolation of thermally stable biradicaloids of the type [E(μ-NTer)]2 (E = P, As; IV in Fig. 1) which can easily be accessed by reduction of [ClE(μ-NTer)]2 with activated magnesium chips.7
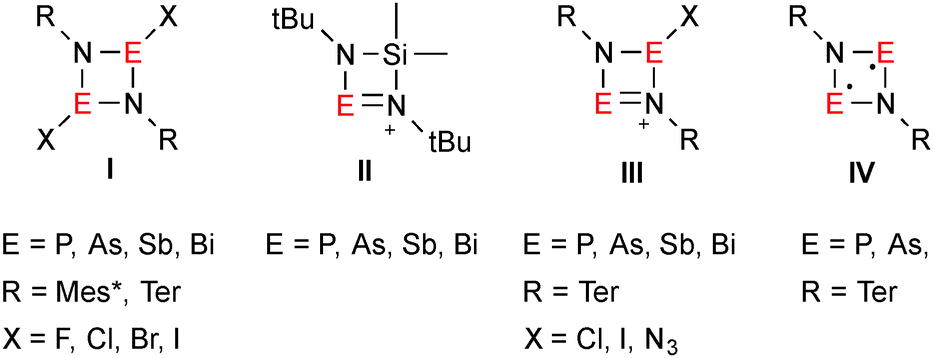 | ||
| Fig. 1 Selected known four-membered E–N heterocycles.4–7 | ||
Just recently, we described the synthesis of stable acyclic chloropnictenium ion salts, with an exceedingly bulky RAr*-group (Ar* = C6H2{C(H)Ph2}2R-2,6,4; R = Me, tBu) attached to the nitrogen atom.8 This sterically demanding moiety offers two flanking phenyl groups for arene-interactions with the low-coordinate reactive site of the molecules. Jones and co-workers realized new bonding situations with the aid of the RAr*-moiety,9 such as mono-coordinate Ge or Sn cations,10 singly bonded distannyene and Ge and Sn hydride complexes,11,12 that showed magnificent activity as a catalyst in hydroboration reactions.13 Just recently, the first example of an amido-distibene in [iPrAr*N(SiiPr3)Sb]2 was reported.14 Herein we describe the synthesis of an unprecedented distannadiazane [Sn(μ-NRAr*)]2 with a planar N2Sn2-core and its trans-metalation with ECl3 (E = Sb, Bi), resulting in the isolation of the first N,N-bis(dichlorostibino)amine and an elusive four-membered ring system with a N2Bi(III)Sn(IV) unit.
In analogy to a procedure described by Power et al., leading to the first isolable distannadiazane [Sn(μ-NTer)]2,15 the exceedingly bulky amine tBuAr*NH2 and Sn{N(SiMe3)2}2 were combined in a Schlenk flask without solvent and heated to 160 °C over a period of 45 min, affording a deep red solid. HN(SiMe3)2 and excess Sn{N(SiMe3)2}2 were removed in vacuo and the crude product was recrystallized from C6H5F to obtain red crystals of [Sn(μ-NtBuAr*)]2 (1R, R = tBu) in moderate yields (64%). The synthesis of 1Me and 1iPr suffered from low solubility of the products in common organic solvents, however, minimal amounts of X-ray quality crystals of 1iPr were obtained from C6H6. In the 13C and 1H NMR spectrum 1iPr and 1tBu can be easily identified by the signals of the para-substituent of the inner phenyl group and their diagnostic 119Sn NMR shifts (1iPr 783.1 ppm, 1tBu 789.2 ppm; cf. [Sn(μ-NTer)]2 738.9 ppm). 1iPr and 1tBu crystallize as solvates of C6H6 or C6H5F (see Fig. S1 and S4 in the ESI†), respectively, in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one molecule in the asymmetric unit, which lies on a crystallographically imposed centre of inversion. In contrast to [Sn(μ-NTer)]2, in which the Sn2N2 ring is characterized by a folding about the Sn⋯Sn axis of 148°, the Sn2N2-core is planar with slightly different N1–Sn1 and N1′–Sn1′ distances (1iPr 2.076(2), 2.086(2); 1tBu 2.075(2), 2.090(2) Å; cf. [Sn(μ-NTer)]2 2.09, 2.11 Å), a transannular Sn1⋯Sn1′ separation of 3.2304(4) (1iPr) and 3.2318(3) Å (1tBu) and rather acute angles at the tin center (1iPr 78.27(7), 1tBu 78.22(6)°, cf. [Sn(μ-NTer)]2 77.6°).15 The nitrogen atoms are in a planar environment as expected for a formal sp2-hybridized center with a p-type lone pair (LP) of electrons. Hence, the planarity of the core is imposed by the increasing bulkiness of the tBuAr*-moieties, as a bend core would result in pyramidalization about the N atoms to fit both RAr*-groups in. Just recently, the bonding in [E(μ-NTer)]2 (E = Ge, Sn, Pb) was studied in detail by Ziegler et al., who analysed the interaction of the monomeric units E(μ-NTer) in the dimeric structure, with the result that the dimer is kept together by two σ- and π-bonds.16
with one molecule in the asymmetric unit, which lies on a crystallographically imposed centre of inversion. In contrast to [Sn(μ-NTer)]2, in which the Sn2N2 ring is characterized by a folding about the Sn⋯Sn axis of 148°, the Sn2N2-core is planar with slightly different N1–Sn1 and N1′–Sn1′ distances (1iPr 2.076(2), 2.086(2); 1tBu 2.075(2), 2.090(2) Å; cf. [Sn(μ-NTer)]2 2.09, 2.11 Å), a transannular Sn1⋯Sn1′ separation of 3.2304(4) (1iPr) and 3.2318(3) Å (1tBu) and rather acute angles at the tin center (1iPr 78.27(7), 1tBu 78.22(6)°, cf. [Sn(μ-NTer)]2 77.6°).15 The nitrogen atoms are in a planar environment as expected for a formal sp2-hybridized center with a p-type lone pair (LP) of electrons. Hence, the planarity of the core is imposed by the increasing bulkiness of the tBuAr*-moieties, as a bend core would result in pyramidalization about the N atoms to fit both RAr*-groups in. Just recently, the bonding in [E(μ-NTer)]2 (E = Ge, Sn, Pb) was studied in detail by Ziegler et al., who analysed the interaction of the monomeric units E(μ-NTer) in the dimeric structure, with the result that the dimer is kept together by two σ- and π-bonds.16
Combining red 1tBu with two equivalents of SbCl3 in CH2Cl2 resulted in an immediate decolourisation, accompanied by a colourless precipitate (Scheme 1, reaction (ii)), which was removed by filtration and from the filtrate X-ray quality crystals of trans-[ClSb(μ-NTer)]2 (2) were grown overnight at room temperature. This metathesis route gives 2 reproducibly in good yields, while using the elimination of SnCl2 as the driving force, which dates back to the seminal work of Veith,17 who established this route to prepare [Me2SiECl(μ-NtBu)2] ring systems (vide supra, Fig. 1 species II).18
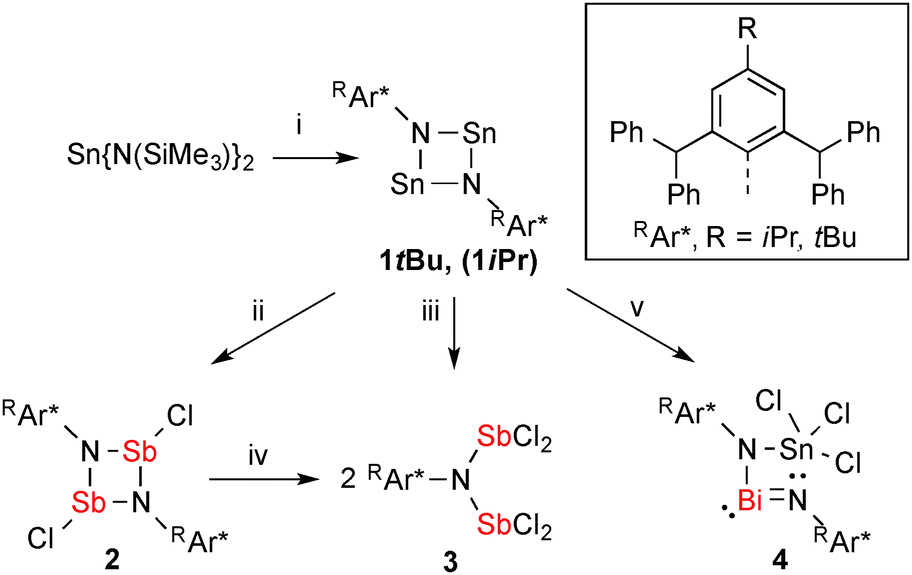 | ||
| Scheme 1 Preparation of 1R-4: (i) 2 RAr*NH2, –2 HN(SiMe3)2, (ii) 2 SbCl3, –2 SnCl2, (iii) 4 SbCl3, –2 SnCl2, (iv) 2 SbCl3, and (v) BiCl3, –Sn. | ||
Pale yellow crystals of 2 are moisture-sensitive, but indefinitely stable in an inert gas atmosphere and can be heated above 270 °C without decomposition. 2 crystallizes solvent-free in the triclinic space group P with one molecule in the unit cell and displays a trans-substituted centrosymmetric dimer with a planar Sb2N2 core protected by two bulky tBuAr* groups similar to the molecular structures of [XSb(μ-NMes*)]2 X = F, Cl, Br, I; trans-[ClSb(μ-NtBu)]2.19,20 As expected the Sb atoms are trigonal pyramidally coordinated, with an s-type LP located on Sb and a trigonal planar coordination environment about the N atom. Additionally, one rather weak dipolar interaction between Sb and a flanking phenyl group (Sb⋯CCt = 3.29 Å, CCt = centroid) is detected (Fig. 2, left).21 The formation of 2 can be reproduced, however, if an excess of SbCl3 is used, a new product tBuAr*N(SbCl2)2 (3) was isolated. Consequently, we reasoned that 3 was accessible directly from 1tBu (reaction (iii) in Scheme 1) when combined with four equiv. of SbCl3, which yielded pure 3. Moreover, treatment of 2 with two additional equiv. of SbCl3 also afforded (reaction (iv) in Scheme 1) 3 in good yields (78%). 3 is thermally stable and melts without decomposition at 236 °C and also shows distinct 1H NMR shifts for the p-tBu, the CHPh2 and the inner phenyl H atoms. Furthermore, 3 belongs to the family of N,N-bis(dichloropnictino)amines, which are well documented for phosphorus (RN(PCl2)2, R = Dipp, Trip, Ph).2 Compound 3 was found to be monoclinic (P21/n) with one molecule of 3 and two disordered C6H5F solvents molecules in the asymmetric unit. The Sb–N distances of 2.030(2) and 2.039(2) Å are shorter than the sum of the covalent radii for Sb and N (cf. ∑rcov(N–Sb) = 2.11 Å)22 representing highly polarized Sb–N single bonds. The trigonal planar N atom lies between both pyramidal SbCl2 units, which adopt a trans configuration with respect to the SbCl2 moieties (Fig. 2 right). Interestingly, two intramolecular Sb⋯Cl contacts (Sb1⋯Cl4, Sb2⋯Cl2 ca. 3.35 Å; cf. ∑rvdW(N–Sb) = 3.81 Å),23 stabilizing this trans configuration, but no intermolecular contacts are observed.
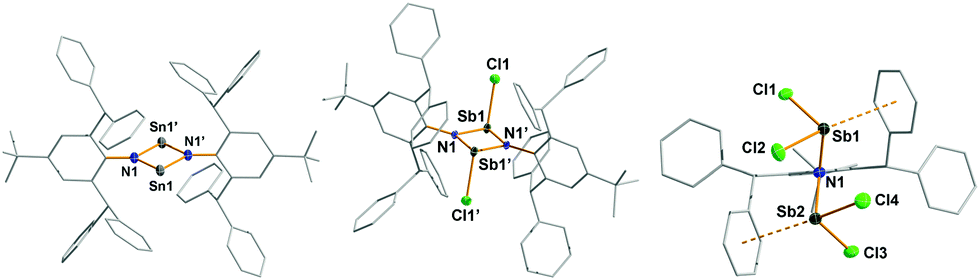 | ||
| Fig. 2 Molecular structures of 1tBu (left), 2 (middle) and 3 (right). Thermal ellipsoids drawn at 50% probability and −100 °C. tBuAr* substituents rendered as wire-frame and H atoms omitted for clarity. Selected bond lengths (Å) and angles (°) of 1tBu: Sn1–N1 2.0752(16), 2.0897(16); N1–Sn1–N1′ 78.22(6); 2: Sb1–N1 2.033(2), Sb1–N1′ 2.034(2), Sb1–Cl1 2.4327(7), Sb1Sb1 3.1749(3), N1–C1 1.430(3) Å, ∑(<Sb) 273.05; ∑(<N) 359.83, C1–C2–N1–Sb1 77.6(2); 3: Sb1–N1 2.030(2), Sb1–Cl1 2.3709(7), Sb1–Cl2 2.4338(7), Sb2–N1 2.039(2), Sb2–Cl3 2.3731(7), Sb2–Cl4 2.4199(7), N1–C1 1.434(3), ∑(<Sb1) 280.08, ∑(<Sb2) 281.47, Sb1–N1–C1–C6 80.0(2). | ||
In addition, the reaction of 1tBu with two equiv. of BiCl3 was studied in CH2Cl2, resulting in a black reaction mixture (reaction (v) in Scheme 1). After multiple filtrations a clear orange solution was obtained. Recrystallization yielded small amounts of orange crystals that were identified as the hitherto unknown [BiSnCl3(μ-NtBuAr*)2] (4). The black residue could not be conclusively identified and we assume that elemental tin is formed in a complex redox process that might also involve the formation of elemental bismuth (vide infra). It has been shown before that the Sn(II) center in [Me2SiSn(μ-NtBu)2] acts as a chloride acceptor in the coupling of phosphaalkenes24 and in the reaction with chlorophosphanes.25
Revision of the reaction conditions prompted us to repeat the experiment in C6H5F with one equivalent of BiCl3 (with respect to 1tBu), to exclude a chloride-shift from CH2Cl2. This again resulted after filtration over a celite-padded frit and concentration of the filtrate in the deposition of orange crystals of 4 as a C6H5F solvate. Only small amounts of pure 4 could be isolated, therefore we cannot provide a comprehensive characterization. Nevertheless, the 119Sn NMR spectrum of these isolated crystals revealed a signal at 115.5 ppm (Fig. S13, ESI†), which is in the expected range for a hypercoordinate N2Sn(IV)Cl3 moiety (cf. Me3SnCl2−: 47.7, Me2SnCl3−: 128 ppm, MeSnCl4−: 274 ppm).26 It should be noted that 119Sn NMR data strongly depend on substitution, coordination number and solvent giving rise to large chemical shift differences (cf. [SnCl3{κ2-DippN(H)C2H4N(Dipp)}] −303 ppm).27 According to MO and NBO analyses of the truncated model [BiSnCl3(μ-NPh)2], 4 can either be described as zwitterionic bismaallyl species (Lewis representation A/C in Fig. 4), as a bismuthenium species (E) or as an iminobismutane (B and D), and therefore represents the first neutral compound with a 4e–3c double bond delocalized along N–Bi–N (Fig. 4). In addition, an s-type lone pair (93%, see Fig. S14 and S15, ESI†) is located at the Bi center. Lewis representations A/C represent the best Lewis structures according to NBO analysis. Along with structures of type E/F, which also possess a rather large weight, since the π bonds are dominantly located at the N atoms (81%), this situation resembles that of N-heterocyclic carbenes (NHC),28 which are stabilized by intramolecular π-donor–π-acceptor interactions (population of the pz(Bi) = 0.47e) to stabilize the dicoordinate carbene C atom. It should be noted that also Bi–N σ bonds (78%) are highly polar, as well as the Sn–Cl or Sn–N bonds (N, Cl: ca. 80%). The computed large positive charges at the Bi and Sn centers are very similar with values of +1.67 and 1.77e supporting the picture of highly polarized Bi–N and Sn–Y (Y = Cl, N) bonds.
4 crystallizes as CH2Cl2 solvate (4·(CH2Cl2)2) in the triclinic space group P with two molecules of 4 and four CH2Cl2 molecules (disordered on their positions) in the cell. Moreover, from C6H5F species 4 crystallizes as a solvate of fluorobenzene solvate (4·C6H5F) in the orthorhombic space group Pna21 (the discussion is led for 4·CH2Cl2). The most prominent structural feature is the planar 4-membered Sn–N–Bi–N heterocycle featuring two different heavy main group metals (deviation from planarity < 2.3°, Fig. 3). Both Bi–N bond lengths are rather short with 2.106(3) and 2.108(3) Å (cf. ∑rcov(N–Bi) = 2.22, (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Bi) = 2.01 Å;22 [Me2SiBi(μ-NtBu)2]+ 2.08 Å, [Bi(IBi)(μ-NTer)2]+ 2.13 Å, and [Me2SiBi(μ-NDipp)2] 2.12 Å, where Dipp = 2,6-iPrC6H3)4,5,29 clearly displaying some Bi–N double bond character in accord with our computation (Fig. 4). Interestingly, both Sn–N bond lengths (2.094(3) and 2.107(3) Å, cf. ∑rcov(N–Sn) = 2.11, (NSn) = 1.90 Å) are in the similar range like the Bi–N distances, however, describing typical highly polarized Sn(IV)–N single bonds. Both the N–Bi–N angle and N–Sn–N angles are rather acute with ca. 78° (cf. [Me2SiBi(μ-NtBu)2]+ 72.9, [Bi(IBi)(μ-NTer)2]+ 77.4°, and [Me2SiBi(μ-NDipp)2]+ 73.7),4,5,29 while the two Bi–N–Sn angles are much larger with 101–102°. A closer look at the secondary interactions revealed that the Sn–N–Bi–N heterocycle is well protected inside the pocket formed by the two tBuAr*-phenyl substituents. However, the dicoordinate bismuth is stabilized by strong secondary interactions (Menshutkin type π complexes)21 with two phenyl groups as indicated by very short Bi⋯centroid distances (2.891/2.978 Å; cf. [MeAr*N(SiMe3)BiCl][Al(ORF)4]+ 2.86/2.94 Å)8 which are well within the range of van-der-Waals radii (∑rvdW(C⋯Bi) = 3.77 Å).23
Bi) = 2.01 Å;22 [Me2SiBi(μ-NtBu)2]+ 2.08 Å, [Bi(IBi)(μ-NTer)2]+ 2.13 Å, and [Me2SiBi(μ-NDipp)2] 2.12 Å, where Dipp = 2,6-iPrC6H3)4,5,29 clearly displaying some Bi–N double bond character in accord with our computation (Fig. 4). Interestingly, both Sn–N bond lengths (2.094(3) and 2.107(3) Å, cf. ∑rcov(N–Sn) = 2.11, (NSn) = 1.90 Å) are in the similar range like the Bi–N distances, however, describing typical highly polarized Sn(IV)–N single bonds. Both the N–Bi–N angle and N–Sn–N angles are rather acute with ca. 78° (cf. [Me2SiBi(μ-NtBu)2]+ 72.9, [Bi(IBi)(μ-NTer)2]+ 77.4°, and [Me2SiBi(μ-NDipp)2]+ 73.7),4,5,29 while the two Bi–N–Sn angles are much larger with 101–102°. A closer look at the secondary interactions revealed that the Sn–N–Bi–N heterocycle is well protected inside the pocket formed by the two tBuAr*-phenyl substituents. However, the dicoordinate bismuth is stabilized by strong secondary interactions (Menshutkin type π complexes)21 with two phenyl groups as indicated by very short Bi⋯centroid distances (2.891/2.978 Å; cf. [MeAr*N(SiMe3)BiCl][Al(ORF)4]+ 2.86/2.94 Å)8 which are well within the range of van-der-Waals radii (∑rvdW(C⋯Bi) = 3.77 Å).23
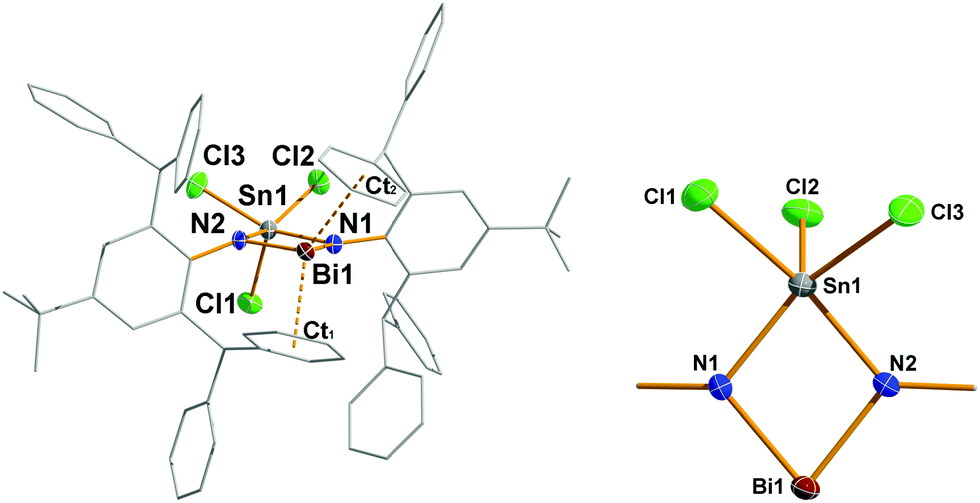 | ||
| Fig. 3 Molecular structures of 4. Thermal ellipsoids drawn at 50% probability and −100 °C. tBuAr* substituents rendered as wire-frame and H atoms omitted for clarity. Selected bond lengths (Å) and angles (°) of 4: Sn1–N1 2.094(3), Sn1–N2 2.107(3), Sn1–Cl1 2.353(1), Sn1–Cl3 2.387(1), Sn1–Cl2 2.403(1), Sn1⋯Bi1 3.2631(4), Bi1–N1 2.106(3), Bi1–N2 2.108(3), N1–C37 1.425(5), N2–C1 1.426(5), N1–Sn1–N2 78.41(12), N1–Bi1–N2 78.10(12), ∑(<N1) 358.0, ∑(<N2) 353.4, Bi1–CCt1 2.891, Bi1–CCt2 2.978 Å. | ||
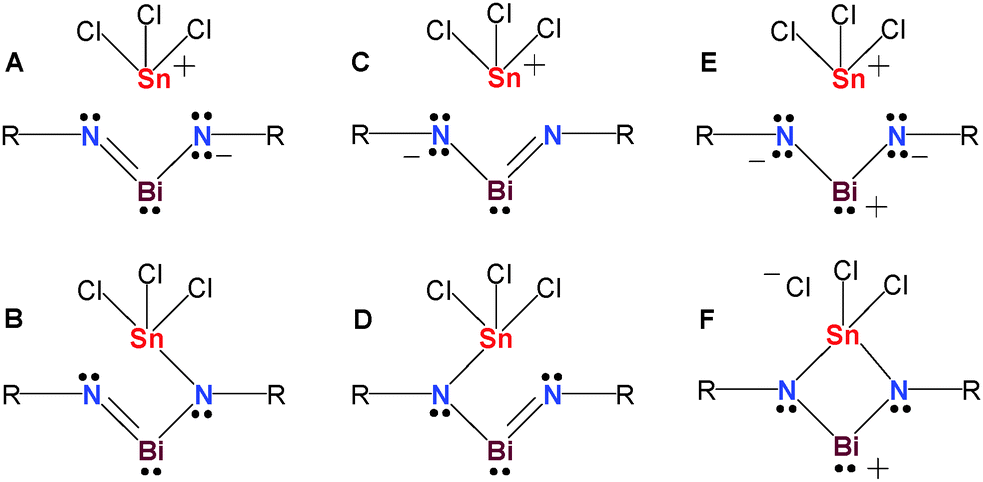 | ||
| Fig. 4 Selected Lewis representations of 4. | ||
In conclusion, we succeeded in the preparation of the first N,N′-bis(dichlorostibinino)amine and an unusual heterocycle containing Sn(IV) and a dicoordinate Bi-center, which is protected by arene-interactions to flanking phenyl groups of the bulky Ar* moiety. These species might be useful starting materials for the preparation of pnictadiazonium salts of Sb and Bi. In comparison to stable N-heterocyclic carbenes,28 the dicoordinated Bi species 4 can be regarded as a heavy atom analog of NHCs.
DFG (SCHU 1170/11-1) is gratefully acknowledged for financial support. C. H.-J. thanks the Fonds der chemischen Industrie for financial support. The authors thank MSc Jonas Bresien for setting up and maintaining Gaussian and NBO software on the cluster computer.
Notes and references
- (a) G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 44, 7815–7880 CrossRef PubMed; (b) M. S. Balakrishna, D. J. Eisler and T. Chivers, Chem. Soc. Rev., 2007, 36, 650–664 RSC.
- (a) F. Reiß, A. Schulz, A. Villinger and N. Weding, Dalton Trans., 2010, 39, 9962 RSC; (b) C. Ganesamoorthy, M. S. Balakrishna, J. T. Mague and H. M. Tuononen, Inorg. Chem., 2008, 47, 7035–7047 CrossRef CAS PubMed; (c) N. Burford, C. T. Stanley, K. D. Conroy, B. Ellis, C. L. B. MacDonald, R. Ovans, A. D. Phillips, P. Ragogna and D. Walsh, Can. J. Chem., 2002, 80, 1404–1409 CrossRef CAS; (d) V. D. Romanenko, A. B. Drapailo, A. N. Chernega and L. N. Markovskii, Zh. Obshch. Khim., 1991, 61, 2434–2441 CAS; (e) S. Goldschmidt and H.-L. Krauß, Liebigs Ann. Chem., 1955, 595, 193–202 CrossRef CAS PubMed.
- D. Michalik, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2010, 46, 7575–7577 ( Angew. Chem. , 2010 , 122 , 7737–7740 ) CrossRef PubMed.
- M. Veith, B. Bertsch and V. Huch, Z. Anorg. Allg. Chem., 1988, 559, 73–88 CrossRef CAS PubMed.
- M. Lehmann, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2012, 51, 8087–8091 ( Angew. Chem. , 2012 , 124 , 8211–8215 ) CrossRef CAS PubMed.
- (a) D. Michalik, A. Schulz, A. Villinger and N. Weding, Angew. Chem., Int. Ed., 2008, 47, 6465–6468 ( Angew. Chem. , 2008 , 120 , 6565–6568 ) CrossRef CAS PubMed; (b) A. Schulz and A. Villinger, Inorg. Chem., 2009, 48, 7359–7367 CrossRef CAS PubMed.
- (a) T. Beweries, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2011, 50, 8974–8978 ( Angew. Chem. , 2011 , 123 , 9136–9140 ) CrossRef CAS PubMed; (b) S. Demeshko, C. Godemann, R. Kuzora, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2013, 52, 2105–2108 ( Angew. Chem. , 2013 , 125 , 2159–2162 ) CrossRef CAS PubMed; (c) A. Hinz, A. Schulz and A. Villinger, Chem. – Eur. J., 2014, 20, 3913–3916 CrossRef CAS PubMed; (d) A. Hinz, R. Kuzora, A. Schulz and A. Villinger, Chem. – Eur. J., 2014, 20, 14659–16673 CrossRef CAS PubMed; (e) A. Hinz, A. Schulz and A. Villinger, Chem. Commun., 2015, 51, 1363–1366 RSC.
- C. Hering-Junghans, M. Thomas, A. Schulz and A. Villinger, Chem. – Eur. J., 2015, 21, 6713–6717 CrossRef CAS PubMed.
- J. Li, A. Stasch, C. Schenk and C. Jones, Dalton Trans., 2011, 40, 10448–10456 RSC.
- J. Li, C. Schenk, F. Winter, H. Scherer, N. Trapp, A. Higelin, S. Keller, R. Pöttgen, I. Krossing and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 9557–9561 ( Angew. Chem. , 2012 , 124 , 9695–9699 ) CrossRef CAS PubMed.
- J. Li, C. Schenk, C. Goedecke, G. Frenking and C. Jones, J. Am. Chem. Soc., 2011, 133, 18622–18625 CrossRef CAS PubMed.
- T. J. Hadlington and C. Jones, Chem. Commun., 2014, 50, 2321 RSC.
- (a) T. J. Hadlington, M. Hermann, J. Li, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2013, 52, 10199–10203 ( Angew. Chem. , 2013 , 125 , 10389–10393 ) CrossRef CAS PubMed; (b) T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028–3031 CrossRef CAS PubMed.
- D. Dange, A. Davey, J. A. B. Abdalla, S. Aldridge and C. Jones, Chem. Commun., 2015, 51, 7128–7131 RSC.
- W. A. Merrill, R. J. Wright, C. S. Stanciu, M. M. Olmstead, J. C. Fettinger and P. P. Power, Inorg. Chem., 2010, 49, 7097–7105 CrossRef CAS PubMed.
- M. Brela, A. Michalak, P. P. Power and T. Ziegler, Inorg. Chem., 2014, 53, 2325–2332 CrossRef CAS PubMed.
- M. Veith, Angew. Chem., Int. Ed. Engl., 1975, 14, 265–266 ( Angew. Chem. , 1975 , 87 , 287–288 ) CrossRef PubMed.
- M. Veith and B. Bertsch, Z. Anorg. Allg. Chem., 1988, 557, 7–22 CrossRef CAS PubMed.
- M. Lehmann, A. Schulz and A. Villinger, Eur. J. Inorg. Chem., 2010, 5501–5508 CrossRef CAS PubMed.
- D. J. Eisler and T. Chivers, Inorg. Chem., 2006, 45, 10734–10742 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Organometallics, 2008, 27, 2361–2395 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- E. Niecke, H. J. Metternich and R. Streubel, Eur. J. Inorg. Chem., 1990, 67–69 CAS.
- J. K. West and L. Stahl, Organometallics, 2012, 31, 2042–2052 CrossRef CAS.
- (a) P. J. Smith and A. P. Tupciauskas, Annu. Rep. NMR Spectrosc., 1978, 8, 291 CrossRef CAS; (b) G. F. Hewitson, Master thesis, Durham University, 1980; (c) J. Ortera, J. Org. Chem., 1981, 221, 57 CrossRef.
- S. M. Mansell, C. A. Russell and D. F. Wass, Dalton Trans., 2015, 44, 9756–9765 RSC.
- (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed; (b) N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed; (c) D. Bézier, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2013, 355, 19–33 CrossRef PubMed.
- R. J. Schwamm, B. M. Day, M. P. Coles and C. M. Fitchett, Inorg. Chem., 2014, 53, 3778 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details and information on X-ray structure elucidation. CCDC 1403993–1403998. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04516a |
| This journal is © The Royal Society of Chemistry 2015 |