
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantio- and diastereoselective synthesis of γ-amino alcohols†
Jorge M. M.
Verkade‡
a,
Peter J. L. M.
Quaedflieg
b,
Gerard K. M.
Verzijl
b,
Laurent
Lefort
b,
Floris L.
van Delft‡
a,
Johannes G.
de Vries
c and
Floris P. J. T.
Rutjes
*a
aInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, NL-6525 AJ Nijmegen, The Netherlands. E-mail: f.rutjes@science.ru.nl
bInnovative Synthesis, DSM ChemTech R&D B.V., PO Box 18, NL-6160 MD Geleen, The Netherlands
cLeibniz-Institut für Katalyse, Universität Rostock, Albert-Einstein-Strasse 29a, 18059 Rostock, Germany
First published on 5th August 2015
Abstract
The γ-amino alcohol structural motif is often encountered in drugs and natural products. We developed two complementary catalytic diastereoselective methods for the synthesis of N-PMP-protected γ-amino alcohols from the corresponding ketones. The anti-products were obtained through Ir-catalyzed asymmetric transfer hydrogenation, the syn-products via Rh-catalyzed asymmetric hydrogenation.
The growing number of enantio- and diastereomerically pure drug candidates has driven the advancement of stereoselective synthetic strategies.1,2 The γ-amino alcohol moiety is often encountered in biologically relevant molecules and hence, general procedures are desired to selectively prepare all of its possible diastereoisomers. Examples of molecules containing the γ-amino alcohol structural motif include the drugs Ritonavir and Lopinavir (both anti-HIV)3 and several 4-hydroxyleucine derivatives (anti-obesity) (Fig. 1).4
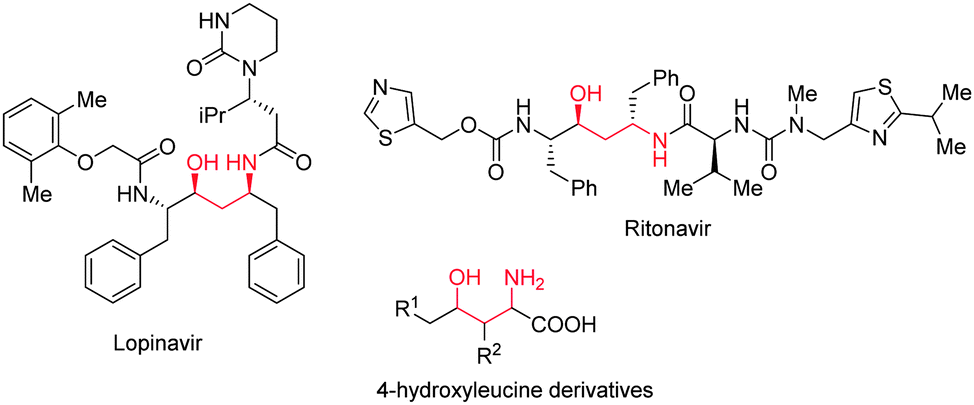 | ||
| Fig. 1 Pharmaceutically relevant γ-amino alcohols. | ||
Despite the abundance of the γ-amino alcohol structure in synthetically relevant targets, relatively few generally applicable stereoselective methods are available for the construction of such a moiety. Undoubtedly the most straightforward route involves diastereoselective reduction of a β-amino ketone Mannich product by employing a suitable hydride donor. Besides several methods for the reduction of α-chiral β-amino ketones,5–7 a number of reports on the stoichiometric reduction of β-branched β-amino ketones (with a methylene adjacent to the amine function) have been disclosed.8–11 These include the diastereoselective reduction of N-sulfonyl-protected γ-hydroxyimines,12syn-selective reductive amination of β-hydroxy ketones with p-anisidine and polymethylhydrosiloxane,13 and dynamic kinetic resolution of N-Boc-protected γ-amino ketones.14 As an alternative, amino alcohols can be prepared through transition metal-catalyzed hydrogenation of β-amino ketones,15 although these methodologies have more generally been reported for the hydrogenation of substances without β-chirality.16
We envisaged that robust enantioselective access to γ-amino alcohols may proceed via a proline-catalyzed Mannich reaction to yield N-PMP-protected amino ketones, diastereoselective reduction of the keto function, and subsequent removal of the PMP protecting group.17 In this report, we describe that N-PMP-protected β-amino ketones can be efficiently converted into each of the corresponding syn- and anti-γ-amino alcohols in a highly diastereoselective manner. Both hydrogenation and transfer hydrogenation have found many applications in stereoselective reduction of alkynes, alkenes, imines and ketones.18 Surprisingly, no literature precedence on the diastereoselective (transfer) hydrogenation of chiral β-amino ketones existed at the start of our research, while on the other hand β-hydroxy ketones have shown to be suitable hydrogenation substrates.19,20 In transfer hydrogenations, 2-propanol or a formic acid/triethylamine mixture is used as the source of hydrogen, which is reversibly transferred to the substrate molecule. Due to this reversibility, a careful analysis of the reaction progress and selectivity is required. We started our investigations on asymmetric transfer hydrogenation (ATH) of N-PMP-protected β-amino ketone 1.
Using the well-established Ru/TsDPEN complex 3 as the catalyst, we observed a clean conversion into the desired γ-amino alcohols with a moderate dr (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20), which irrespective of the existing chiral center depended on the catalyst chirality (Scheme 1). Encouraged by these initial results we also explored the use of iridium-based ATH catalysts. We prepared catalysts 4 by heating a solution of a suitable iridium precursor (i.e. [IrCp*Cl2]2) and an amino acid amide in the presence of an inorganic base (e.g. K2CO3) according to a modified protocol disclosed by Verzijl.21 The inorganic base was removed by filtration to suppress possible elimination of p-anisidine prior to reduction. Preferably, α,α-disubstituted amino acids were employed to avoid the risk of catalyst racemization.
:20), which irrespective of the existing chiral center depended on the catalyst chirality (Scheme 1). Encouraged by these initial results we also explored the use of iridium-based ATH catalysts. We prepared catalysts 4 by heating a solution of a suitable iridium precursor (i.e. [IrCp*Cl2]2) and an amino acid amide in the presence of an inorganic base (e.g. K2CO3) according to a modified protocol disclosed by Verzijl.21 The inorganic base was removed by filtration to suppress possible elimination of p-anisidine prior to reduction. Preferably, α,α-disubstituted amino acids were employed to avoid the risk of catalyst racemization.
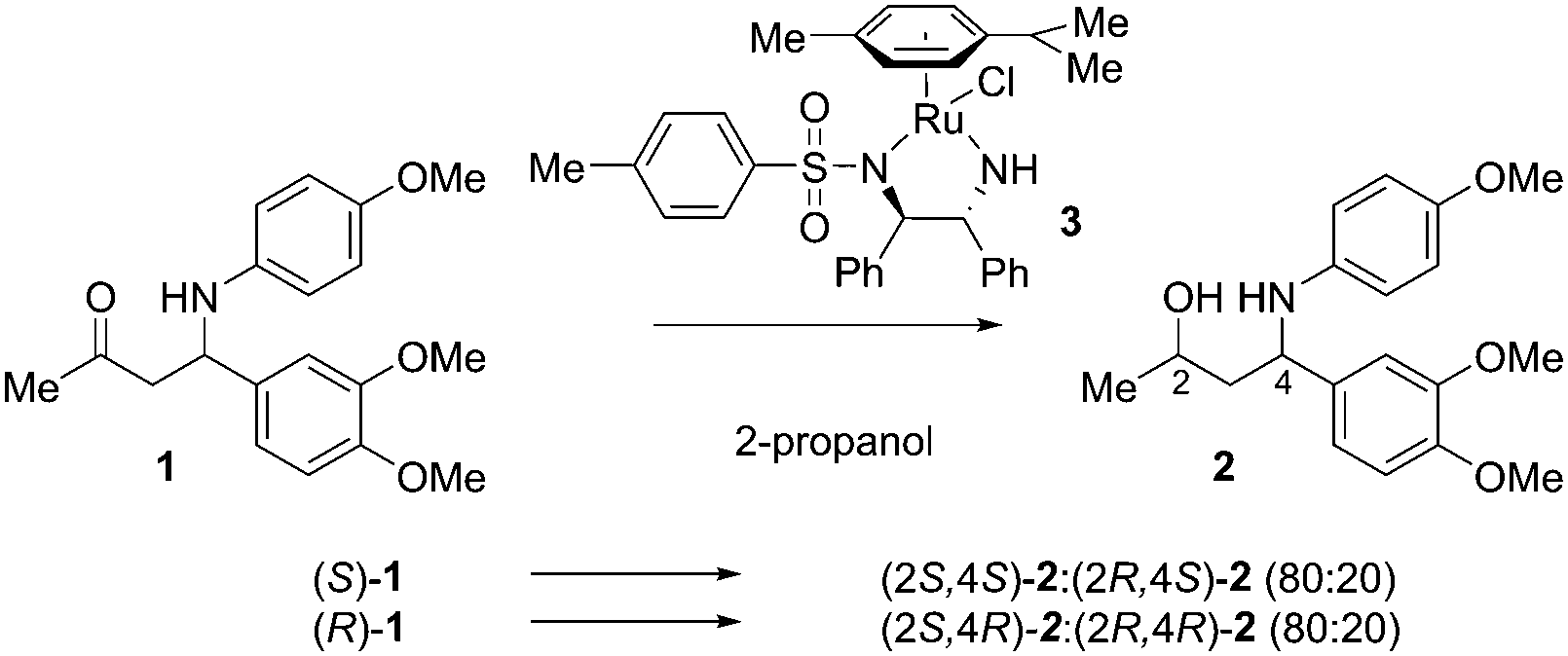 | ||
| Scheme 1 Ru/(R,R)-TsDPEN-catalyzed ATH of ketone 1. | ||
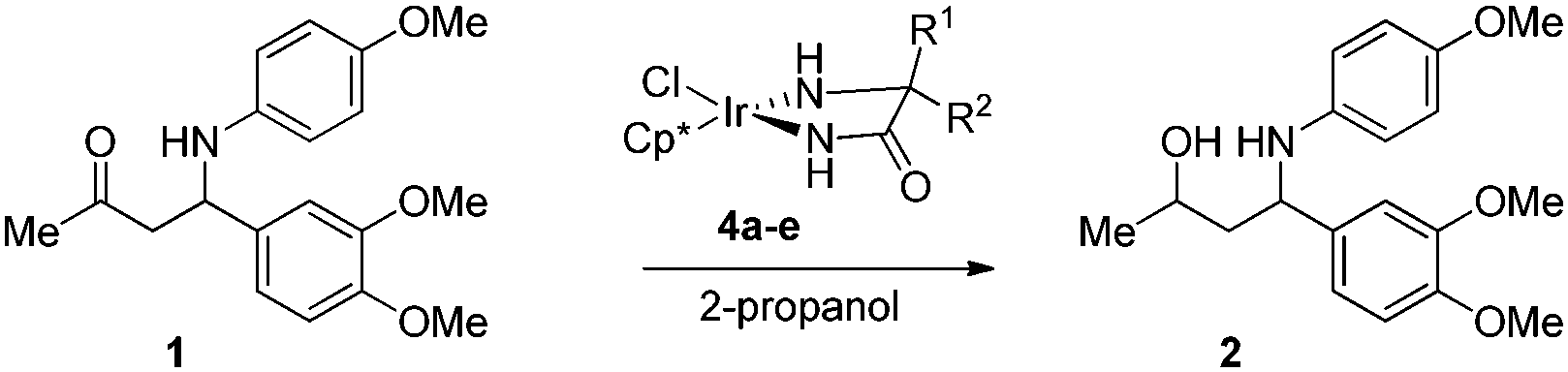
To our satisfaction, exposure of benchmark substrate 1 to these catalysts resulted in high diastereoselectivities. When D-α-Me-phenylglycine amide was used as the ligand, conversion of (S)-1 into the corresponding anti-amino alcohol 2 proceeded in a diastereomeric ratio of 96:4 (Table 1, entry 1), while the (R)-aminoketone led to a 1:1 formation of amino alcohols (entry 2). This implies that during iridium-catalyzed reduction, the existing chiral center has a large impact on the stereochemical outcome of the transfer hydrogenation. The influence of the preexisting chirality in terms of a match and mismatch with the ligand was confirmed by employing achiral Aib-NH2 as the ligand (entry 3). In the presence of this achiral catalyst, a diastereomeric ratio of 84:16 was observed for the products. Replacing substituent R2 of catalyst 4a with a Bn group (i.e.4d) resulted in decreased selectivity (entry 4), whereas nearly complete selectivity was obtained with the same catalyst 4d for the (R)-substrate (entry 5). The combination of phenyl and benzyl substituents showed again a clear match (entry 6, diastereoselectivity of 0:100) and mismatch (entry 7).
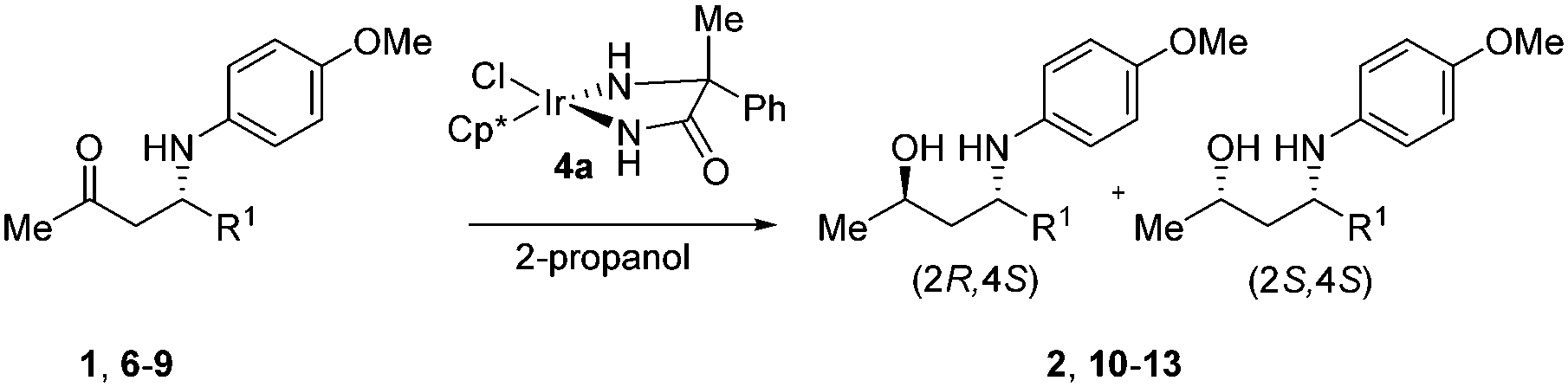
Although slightly better results were obtained with α-benzylated phenylglycinamide as the ligand, we explored the substrate scope of the stereoselective ATH with the α-methyl-α-phenyl substituted glycinamide-based catalyst (4a) because of its straightforward accessibility. The β-amino ketone substrates were prepared via the asymmetric proline-catalyzed Mannich reaction.22,23 The results in Table 2 led us to conclude that ATH of β-amino ketones is widely applicable. In all examples we observed a reasonable to good diastereoselectivity, with the best selectivities obtained for R1 = Ar. In addition, it is worth mentioning that we have previously successfully deprotected both diastereoisomers of PMP-protected amino alcohol 2 using oxidative enzymatic conditions.17b
|
|
|||||
|---|---|---|---|---|---|
| Entry | sm | R1 | pr | drb | Yieldc |
|
a Reaction conditions: ketone (1.0 equiv.), (IrCp*Cl2)2 (0.02 equiv.), α-Me-phenylglycine-NH2 (0.20 equiv.), K2CO3 (3 equiv.), 2-propanol, rt, 1.5–20 h.
b (2R,4S):(2S,4S) (determined by HPLC).
c Isolated yield.
d Absolute configuration = (2R,4R):(2S,4R).
|
|||||
| 1 | (S)-1 | 3,4-(MeO)2C6H3 | 2 | 96:4 |
100 |
| 2 | (S)-6 | 4-FC5H4 | 10 | 95:5 |
88 |
| 3 | (S)-7 | 2-MeC6H4 | 11 | 97:3 |
76 |
| 4 | (R)-8 | iBu | 12 | 76:24d |
99 |
| 5 | (S)-9 | CO2Et | 13 | 79:21 |
100 |
With an efficient method for the anti-selective preparation of γ-amino alcohols in hand, we realized that extensive screening of other metal/ligand combinations could possibly deliver γ-amino alcohols with syn-selectivity. We nevertheless resorted to hydrogenation with molecular hydrogen for the synthesis of the syn-congeners. We discovered that hydrogenation of β-amino ketones in the presence of a catalyst in situ prepared from Rh(COD)2BF4 and a C2-symmetric ligand such as (R)-BINAP (5) (Table 3), produced the desired syn-γ-amino alcohols with excellent diastereoselectivity.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | sm | R1 | t (h) | pr | drb | Yieldc |
|
a Reaction conditions: substrate (1.0 equiv.), Rh(COD)2BF4 (0.05c equiv.), (R)-BINAP (0.05c equiv.), r.t., 15–44 h or substrate (1.0 equiv.), Rh(COD)2BF4 (0.30d equiv.), (R)-BINAP (0.030 equiv.), 50 °C, 15–44 h.
b (2S,4S):(2R,4S) (determined by HPLC).
c Isolated yields.
d 50 °C.
e rt.
f (2S,4R):(2R,4R) (determined by HPLC).
|
||||||
| 1 | (S)-1 | 3,4-(MeO)2C6H3 | 19d | 2 | >95:5 |
77 |
| 2 | (S)-6 | 4-FC5H4 | 44e | 10 | >95:5 |
76 |
| 3 | (S)-7 | 2-MeC6H4 | 44e | 11 | >95:5 |
81 |
| 4 | (R)-8 | iBu | 15d | 12 | >95:5f |
77 |
| 5 | (S)-9 | CO2Et | 17d | 13 | >95:5 |
56 |
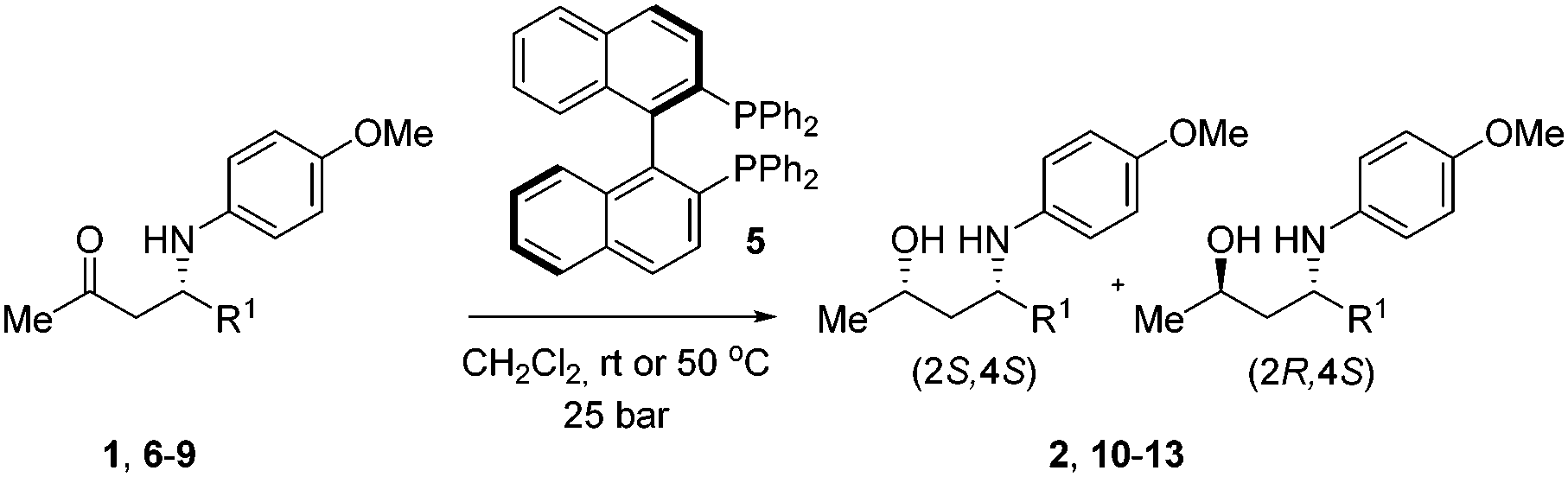
Again we observed a strong effect of the existing chiral center on the diastereoselectivity. Upon hydrogenation of (S)-1 with Rh/(R)-BINAP, pure (2S,4S)-2 was obtained, whereas with Rh/(S)-BINAP the ratio (R,S) vs. (S,S) was 70:30. Dichloromethane appeared to be the most suitable solvent with respect to solubility of the starting material, diastereoselectivity and reaction rate. To investigate the scope and limitations, we subsequently hydrogenated a number of aromatic, aliphatic and carboxylic β-aminoketones on preparative scale (Table 3).
In some cases, the reactions proceeded somewhat slowly, despite the use of higher catalyst loadings (entries 2 and 3). In all cases, however, nearly exclusive formation of the desired syn-diastereoisomer was observed in combination with good yields.
Finally, to verify the assigned stereochemical outcome, we prepared (2S,4S)-2 on a larger scale, after which X-ray crystallographic analysis of the product proved that Rh/(R)-BINAP (5) hydrogenation of (S)-1 indeed led to formation of the syn-product ((2S,4S)-2, Fig. 2).
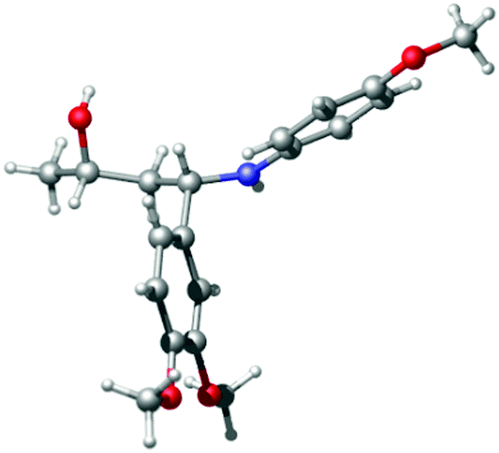 | ||
| Fig. 2 Crystal structure representation of (2S,4S)-2 (ORTEP probability level 50%).24 | ||
We have developed two complementary methods for the hydrogenation of β-amino ketones to the corresponding γ-amino alcohols. The anti-products can be obtained through ATH, in which 2-propanol is employed as the hydrogen donor and an Ir/α-substituted-amino acid amide complex as the catalyst. syn-Products are accessible by asymmetric hydrogenation under hydrogen pressure in the presence of a Rh-based BINAP catalyst. In combination with the proline-catalyzed Mannich reaction, these methods provide powerful tools for the enantio- and diastereoselective synthesis of all four diastereomers of γ-amino alcohols.
This work forms part of the Ultimate Chiral Technology project supported in part with funds provided by SNN (Cooperation Northern Netherlands) and EFRD (European Fund for Regional Development).
Notes and references
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788 CrossRef CAS PubMed.
- H.-J. Federsel, Nat. Rev. Drug Discovery, 2005, 4, 685–697 CrossRef CAS PubMed.
- E. dos Santos Pinheiro, O. Augusto Ceva Antunes and J. M. D. Fortunak, Antiviral Res., 2008, 79, 143 CrossRef PubMed.
- L. Jette, P. McNicol, M. Gill and A. Marette, WO2007107008 A1 2007, compounds and compositions for use in the prevention and treatment of disorders of fat metabolism and obesity.
- G. Bartoli, M. Bartolacci, M. Cortese, E. Marcantoni, M. Massaccesi, R. Pela and L. Sambri, Eur. J. Org. Chem., 2004, 2359 CrossRef CAS PubMed.
- J. Barluenga, E. Aguilar, S. Fustero, B. Olano and A. L. Viado, J. Org. Chem., 1992, 57, 1219 CrossRef CAS.
- Y. Hayashi, T. Urushima, M. Shin and M. Shoji, Tetrahedron, 2005, 61, 11393 CrossRef CAS PubMed.
- R. A. Pilli, D. Russowsky and L. A. Dias, J. Chem. Soc., Perkin Trans. 1, 1990, 1213 RSC.
- D. Berkeš, A. Kolarovič and F. Považanec, Tetrahedron Lett., 2000, 41, 5257 CrossRef.
- G. Keck and P. Truong, Org. Lett., 2002, 4, 3131 CrossRef CAS PubMed.
- F. A. Davis, P. M. Gaspari, B. M. Nolt and P. Xu, J. Org. Chem., 2008, 73, 9619 CrossRef CAS PubMed.
- T. Kochi, T. P. Tang and J. A. Ellmann, J. Am. Chem. Soc., 2003, 125, 11276 CrossRef CAS PubMed.
- D. Menche, F. Arikan, J. Li and S. Rudolph, Org. Lett., 2007, 9, 267 CrossRef CAS PubMed.
- R. Millet, A. M. Träff, M. L. Petrus and J.-E. Bäckvall, J. Am. Chem. Soc., 2010, 132, 15182 CrossRef CAS PubMed.
- Q. Hu; Z. Zhang, Y. Liu, T. Imamoto and W. Zhang, Angew. Chem., Int. Ed., 2015, 54, 2260 CrossRef PubMed.
- (a) K. Mashima, K.-h. Kusano, N. Sato, Y.-i. Matsumura, K. Nozaki, H. Kumobayashi, N. Sayo, Y. Hori, T. Ishizaki, S. Akutagawa and H. Takaya, J. Org. Chem., 1994, 59, 3064 CrossRef CAS; (b) T. Ohkuma, D. Ishii, H. Takeno and R. Noyori, J. Am. Chem. Soc., 2000, 122, 6510 CrossRef CAS; (c) T. Ohkuma, M. Koizumi, K. Muiz, G. Hilt, C. Kabuto and R. Noyori, J. Am. Chem. Soc., 2002, 124, 6508 CrossRef CAS PubMed; (d) D. G. Genov and D. J. Ager, Angew. Chem., Int. Ed., 2004, 43, 2816 CrossRef CAS PubMed; (e) H. L. Ngo and W. Lin, J. Org. Chem., 2005, 70, 1177 CrossRef CAS PubMed; (f) Q. Jing, X. Zhang, J. Sun and K. Ding, Adv. Synth. Catal., 2005, 347, 1193 CrossRef CAS PubMed; (g) D. Liu, W. Gao, C. Wang and X. Zhang, Angew. Chem., Int. Ed., 2005, 44, 1687 CrossRef CAS PubMed; (h) H. Takahashi, S. Sakuraba, H. Takeda and K. Achiwa, J. Am. Chem. Soc., 1990, 112, 5876 CrossRef CAS; (i) S. Sakurabi and K. Achiwa, Synlett, 1991, 689 CrossRef; (j) M. Devocelle, F. Agbossou and A. Mortreux, Synlett, 1997, 1306 CrossRef.
- (a) J. M. M. Verkade, L. J. C. van Hemert, P. J. L. M. Quaedflieg, P. L. Alsters, F. L. van Delft and F. P. J. T. Rutjes, Tetrahedron Lett., 2006, 47, 8109 CrossRef CAS PubMed; (b) J. M. M. Verkade, L. J. C. van Hemert, P. J. L. M. Quaedflieg, H. E. Schoemaker, M. Schürmann, F. L. van Delft and F. P. J. T. Rutjes, Adv. Synth. Catal., 2007, 349, 1332 CrossRef CAS PubMed.
- Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed.
- For a review on the stereoselective synthesis of 1,3-diols, including (transfer) hydrogenation of 3-hydroxyketones, see: S. E. Bode, M. Wolberg and M. Müller, Synthesis, 2006, 557–558 CAS.
- (a) O. Labeeuw, C. Roche, P. Phansavath and J.-P. Genêt, Org. Lett., 2007, 9, 105 CrossRef CAS PubMed; (b) C. Roche, O. Labeeuw, M. Haddad, T. Ayad, J.-P. Genet, V. Ratovelomanana-Vidal and P. Phansavath, Eur. J. Org. Chem., 2009, 3977 CrossRef CAS PubMed.
- G. K. M. Verzijl, NL1009346 (C2), 1998, Asymmetrische transferhydrogering.
- B. List, J. Am. Chem. Soc., 2000, 122, 9336 CrossRef CAS.
- It should be noted that in some instances, Mannich ketones were prone to partial racemization over time. We hypothesize that racemization occurs via catalysis by trace impurities in the Mannich product samples, because ketones 6–9 were oils and purified by troublesome column chromatography, while 1 was purified through crystallization and the resulting crystals appeared more stable.
- xyz file generated with Mercury 3.5.1 (http://www.ccdc.cam.ac.uk/Solutions/CSDSystem/Pages/Mercury.aspx). Picture generated with CYLview, 1.0b; C. Y. Legault, Université de Sherbrooke, 2009 (http://www.cylview.org). CCDC 1400959.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental data, characterization data and crystal data and structure refinements. CCDC 1400959. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04445f |
| ‡ Present address: Synaffix BV, Pivot Park, Molenstraat 110, 5342 CC Oss, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2015 |