
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Programmed assembly of 4,2′:6′,4′′-terpyridine derivatives into porous, on-surface networks†
Thomas
Nijs
a,
Frederik J.
Malzner
b,
Shadi
Fatayer
ac,
Aneliia
Wäckerlin
a,
Sylwia
Nowakowska
a,
Edwin C.
Constable
b,
Catherine E.
Housecroft
*b and
Thomas A.
Jung
*d
aDepartment of Physics, University of Basel, Klingelbergstrasse 82, 4056 Basel, Switzerland
bDepartment of Chemistry, University of Basel, Spitalstrasse 51, 4056 Basel, Switzerland. E-mail: catherine.housecroft@unibas.ch
cInstituto de Física “Gleb Wataghin”, Universidade Estadual de Campinas, Rua Sérgio Buarque de Holanda 777, 13083-859 Campinas, SP, Brazil
dLaboratory for Micro- and Nanotechnology, Paul Scherrer Institut, 5232 Villigen, Switzerland. E-mail: thomas.jung@psi.ch
First published on 3rd July 2015
Abstract
The use of divergent, V-shaped, 4,2′:6′,4′′-terpyridine building blocks that self-assemble into hydrogen-bonded domains and upon addition of copper atoms undergo metallation with concomitant transformation into a coordination network is described; multiple energetically similar structural motifs are observed in both hydrogen-bonded and adatom-coordinated networks.
The assembly of 2-dimensional metal–organic networks on surfaces is topical1 in view of their relationship to 3-dimensional metal–organic frameworks (MOFs).2 However, the mechanism of their assembly (co-determined by the proximity of the surfaces and the presence of adatoms) remains sparsely investigated. Examples of systems which exhibit on-surface association through hydrogen bonding or metal coordination using well defined and controllable motifs include 4,9-diaminoperylene-quinone-3,10-diimine, helicenes and porphyrins.3–8
In contrast to the chelating ligand 2,2′:6′,2′′-terpyridine (2,2′:6′,2′′-tpy),9 4,2′:6′,4′′-terpyridine (4,2′:6′,4′′-tpy) coordinates through only two N atoms and defines a divergent V-shaped building-block allowing control over the assembly of coordination polymers and networks.10 Part of the attractiveness of 4,2′:6′,4′′-tpy ligands in supramolecular chemistry is the simplicity of the synthetic routes11 to 4′-aryl functionalized derivatives (Scheme 1) which allows facile structural and electronic tuning. On-surface investigations of terpyridines are limited. Scanning tunnelling microscopy (STM) shows that 2,2′:6′,2′′-tpy solution-cast onto Au(111) adsorbs with molecules oriented orthogonally to the surface with dominant intermolecular π-stacking.12 A variety of STM studies of adsorbed functionalized 2,2′:6′,2′′-tpys, of adsorbed [M(2,2′:6′,2′′-tpy)2]n+ complexes, and of metal coordination-driven assemblies on either highly ordered pyrolytic graphite (HOPG), Pt(111), Au(111) or Cu(100) have been reported,13 and a recent publication reveals the influence that solvent has in directing surface assemblies from drop-cast films of 1,16-bis([2,2′:6′,2′′-terpyridin]-4′-yloxy)hexadecane.14 However, on-surface assemblies of 4,2′:6′,4′′-terpyridines and their metal complexes remain unexplored.
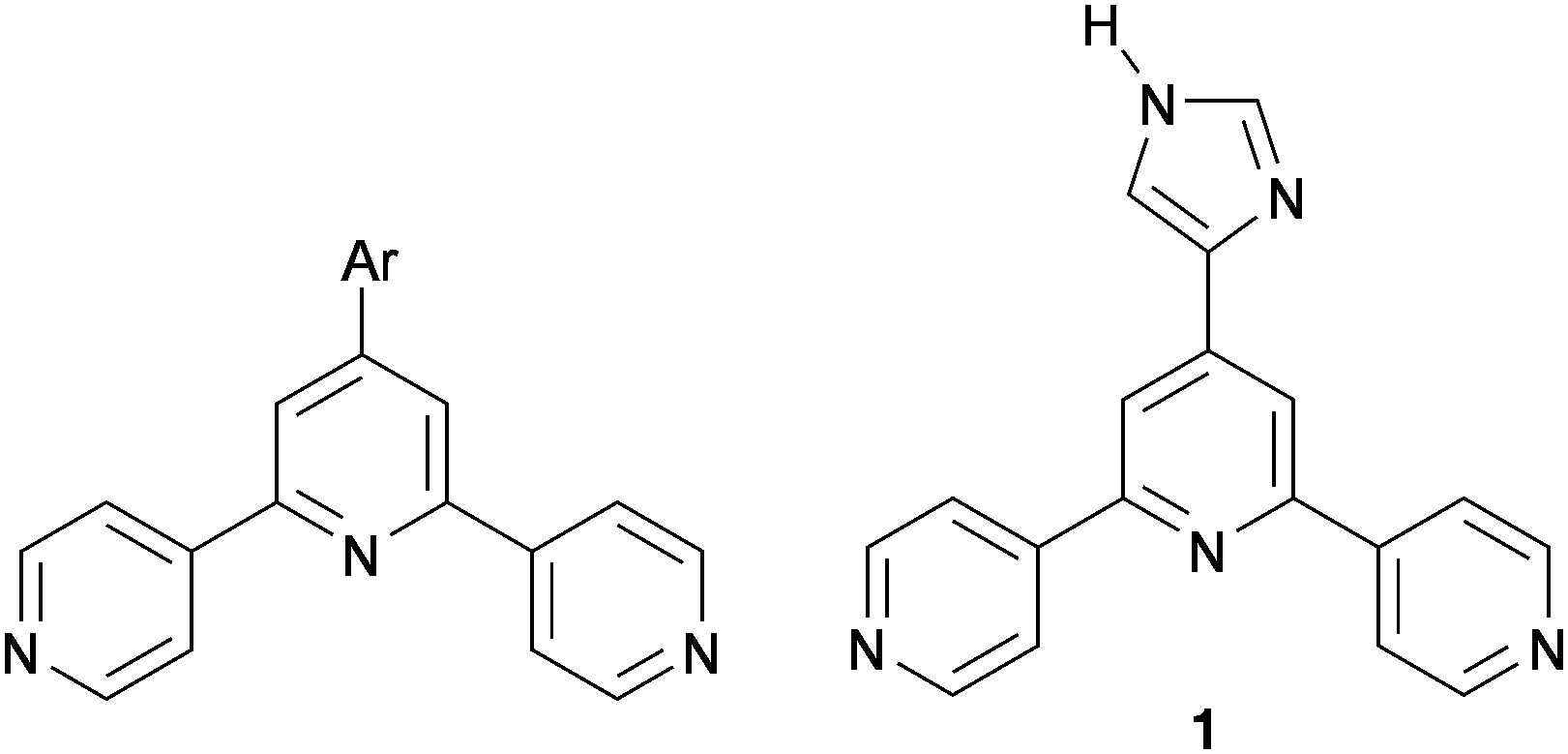 | ||
| Scheme 1 General 4′-aryl-functionalized 4,2′:6′,4′′-terpyridine and the imidazole-functionalized ligand 1. | ||
We present here an investigation of the imidazolyl-functionalized derivative 1 (Scheme 1) on Au(111) and the effects of the addition of copper adatoms. We have previously reported the solid-state structure of 1 CHCl3 and showed that NHimidazole⋯Ntpy hydrogen bonds are favoured over NHimidazole⋯Nimidazole interactions, consistent with the relative basicities of the heterocycles.15 The solid-state structure of only one complex of 1 has been described; in [{2Co(1)2(NCS)2·5H2O}n], the imidazole domain and the central N atom of the 4,2′:6′,4′′-tpy unit are not coordinated.15 A feature of 1 relevant to on-surface assembly is the fact that it is prochiral.
After deposition on an Au(111) substrate, 1 self-assembles into a close-packed phase (Fig. 1b and c) which co-exists with a regular 6-fold nanoporous structure (Fig. 1d and e); between the two phases lies a domain with an irregular assembly pattern (Fig. 1a). The two phases can be rationalized in terms of different intermolecular hydrogen-bonding patterns. The co-existence of two phases is reproducible in different samples and is consistent with a small energy difference between the 2-dimensional assembly motifs.
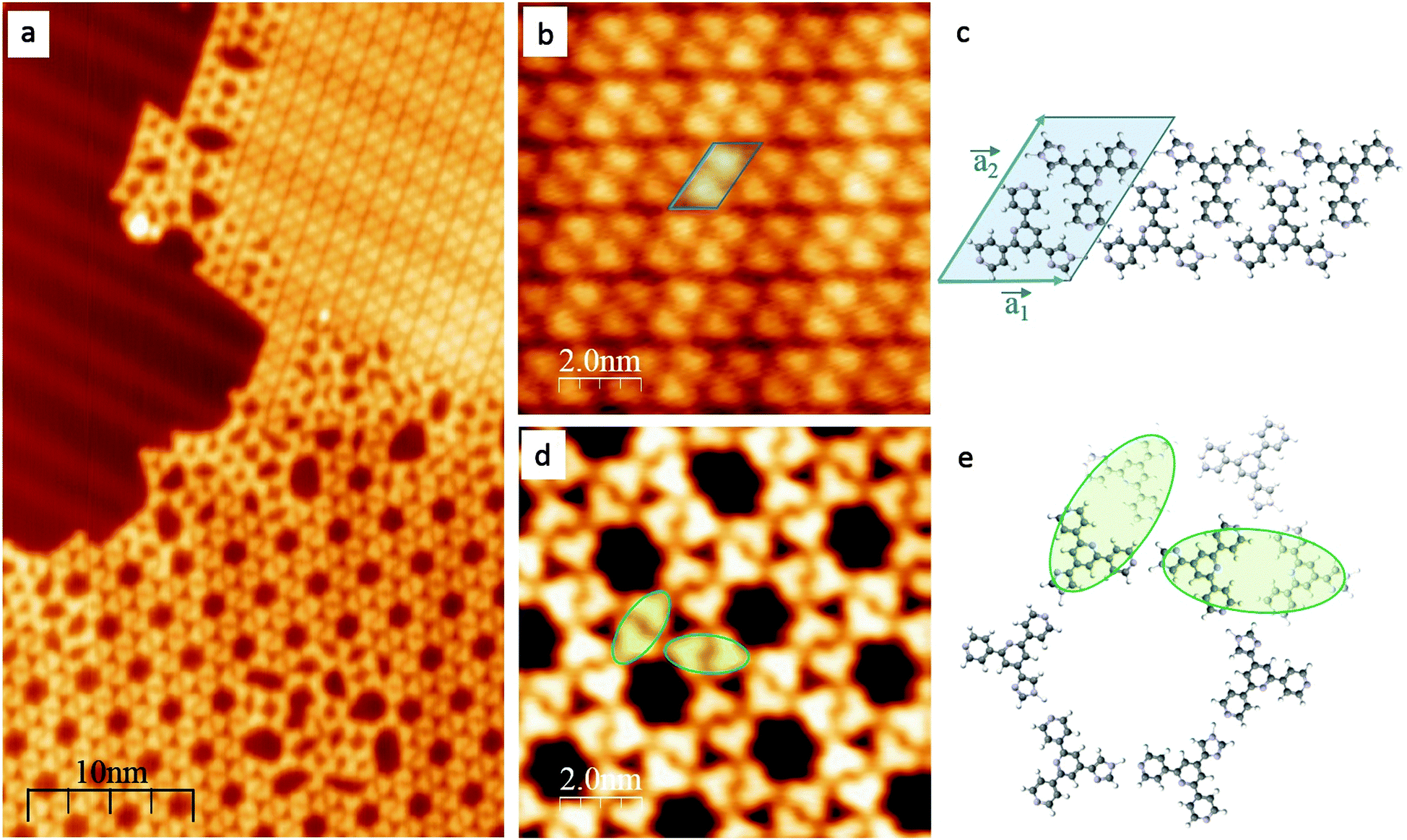 | ||
| Fig. 1 STM images of hydrogen bonded structures of 1 on Au(111). (a) STM image of two coexisting phases: on top, a close-packed array, at the bottom a network structure. Note the irregular assemblies where the two phases meet. (b) STM image of the close-packed phase composed of molecular rows that consists of dimers shown in (c). (c) Model for the adsorption of molecules presents a rhombic shape with lattice constants equal to a1 = (1.21 ± 0.06) nm, a2 = (1.95 ± 0.06) nm and α = (56 ± 2)°. (d) STM image of the hexagonal porous network. The distance between pores of the network is (3.09 ± 0.03)nm and the diameter of the pores is (1.38 ± 0.06) nm which gives an area of (1.52 ± 0.08) nm2. (e) Model of a pore in the network with three adjacent molecules (see d); two dimer motifs are highlighted to illustrate part of a chiral ‘flower’ pattern that runs around the macrocycle. | ||
The first compact assembly (Fig. 1b) consists of a hydrogen-bonded linear double row arrangement, each of these separated by a small gap, due to resulting repulsive interactions (Fig. S1†). The principal motif in the porous assembly is a hexameric array. This is well-modelled by molecules of 1 engaging in NHimidazole⋯Ntpy hydrogen bonding and forming a chiral motif (Fig. 1e and Fig. S2†). All the hexamers in the domain possess the same handedness and the relationship between adjacent cyclic motifs can be seen by defining the dimer shown in Fig. 1e and Fig. S2.† Domains with opposite handedness are present on the surface (Fig. S3†). The hydrogen-bonded supramolecular arrangement of 1 is not significantly influenced by the atomic lattice of the underlying substrate which bears the 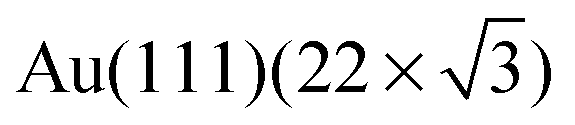 reconstruction.16 The weak corrugation of the supramolecular layer can be attributed to the stacking fault zones of that same reconstruction, visible in both the close-packed and nanoporous phases, and possesses comparable periodicity. This observation of coexisting compact and porous 2-dimensional assemblies is not unique, and of particular relevance are results from Reichert et al.6 who have described a prochiral carbonitrile derivative assembling on Ag(111) in coexisting dense and enantiopure porous phases. The assembly shown in Fig. 1d is similar to a Kagome lattice;17,18 the latter comprises a regular network of interconnected hexagons and triangles, whereas in Fig. 1d, closer association of the hexagonal motifs leads to a reduction in the triangular domains.
reconstruction.16 The weak corrugation of the supramolecular layer can be attributed to the stacking fault zones of that same reconstruction, visible in both the close-packed and nanoporous phases, and possesses comparable periodicity. This observation of coexisting compact and porous 2-dimensional assemblies is not unique, and of particular relevance are results from Reichert et al.6 who have described a prochiral carbonitrile derivative assembling on Ag(111) in coexisting dense and enantiopure porous phases. The assembly shown in Fig. 1d is similar to a Kagome lattice;17,18 the latter comprises a regular network of interconnected hexagons and triangles, whereas in Fig. 1d, closer association of the hexagonal motifs leads to a reduction in the triangular domains.
After sublimation of Cu onto the hydrogen-bonded assemblies of 1 on Au(111), the supramolecular adlayer changes its structure. Chains of linked heterocyclic macrocycles, which generally follow the fcc domains of the 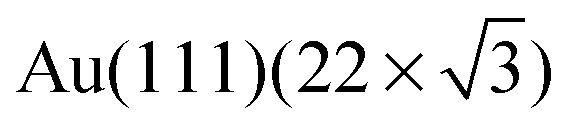 reconstruction, are clearly observed by STM (Fig. 2a). These mostly form regular polygons. The transition upon adding Cu adatoms from extended networks with pseudo-hexagonal symmetry showing little distortion by the Au(111) surface reconstruction, to the oligomeric structures with a preference for the fcc domains of the reconstructed surface, suggests a modified substrate–adsorbate interaction. Such behaviour is plausible for molecular modules coordinated via metal adatoms which are in registry with the substrate atoms. In addition to the STM topographs recorded at 5 K, the local chemical transitions were also analysed by X-ray Photoelectron Spectroscopy (XPS) measurements. These confirm metal coordination by investigating the N 1s binding energies of 1 (Fig. 2b and Table S1†). Prior to coordination, two peaks are observed; the higher energy peak corresponds to NHimidazole (399.6 eV, green in Fig. 2b) and the lower to –N
reconstruction, are clearly observed by STM (Fig. 2a). These mostly form regular polygons. The transition upon adding Cu adatoms from extended networks with pseudo-hexagonal symmetry showing little distortion by the Au(111) surface reconstruction, to the oligomeric structures with a preference for the fcc domains of the reconstructed surface, suggests a modified substrate–adsorbate interaction. Such behaviour is plausible for molecular modules coordinated via metal adatoms which are in registry with the substrate atoms. In addition to the STM topographs recorded at 5 K, the local chemical transitions were also analysed by X-ray Photoelectron Spectroscopy (XPS) measurements. These confirm metal coordination by investigating the N 1s binding energies of 1 (Fig. 2b and Table S1†). Prior to coordination, two peaks are observed; the higher energy peak corresponds to NHimidazole (399.6 eV, green in Fig. 2b) and the lower to –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) (398.3 eV, red in Fig. 2b, both tpy and imidazole) with a well-fitting energy difference of 1.3 eV.19,20 Instead of the ratio being 1
(398.3 eV, red in Fig. 2b, both tpy and imidazole) with a well-fitting energy difference of 1.3 eV.19,20 Instead of the ratio being 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 corresponding to the structure of 1, it is closer to 2:3. This could be due to surface charge effects and/or intermolecular hydrogen bonds,21 as the multilayer shows 1:4 stoichiometry with corresponding peak positions of 400.3 eV respectively 398.9 eV (Fig. S5†). As stated earlier, NHimidazole and the central Ntpy of 1 remain uncoordinated upon Cu-adatom addition and this is consistent with the XPS peaks shown in green and red in Fig. 2b which remain at the same peak positions (Fig. 2bversusFig. 2c). The second Nimidazole and the two outer pyridine Ntpy undergo coordination which is confirmed by a shift of 1.4 eV to higher binding energy to 399.7 eV (orange in Fig. 2c).22 The ratio of peak areas (orange:green:red in Fig. 2c) was set to be 2:1:1. The binding energies for uncoordinated and coordinated molecules are summarized in Table S1.†
:4 corresponding to the structure of 1, it is closer to 2:3. This could be due to surface charge effects and/or intermolecular hydrogen bonds,21 as the multilayer shows 1:4 stoichiometry with corresponding peak positions of 400.3 eV respectively 398.9 eV (Fig. S5†). As stated earlier, NHimidazole and the central Ntpy of 1 remain uncoordinated upon Cu-adatom addition and this is consistent with the XPS peaks shown in green and red in Fig. 2b which remain at the same peak positions (Fig. 2bversusFig. 2c). The second Nimidazole and the two outer pyridine Ntpy undergo coordination which is confirmed by a shift of 1.4 eV to higher binding energy to 399.7 eV (orange in Fig. 2c).22 The ratio of peak areas (orange:green:red in Fig. 2c) was set to be 2:1:1. The binding energies for uncoordinated and coordinated molecules are summarized in Table S1.†
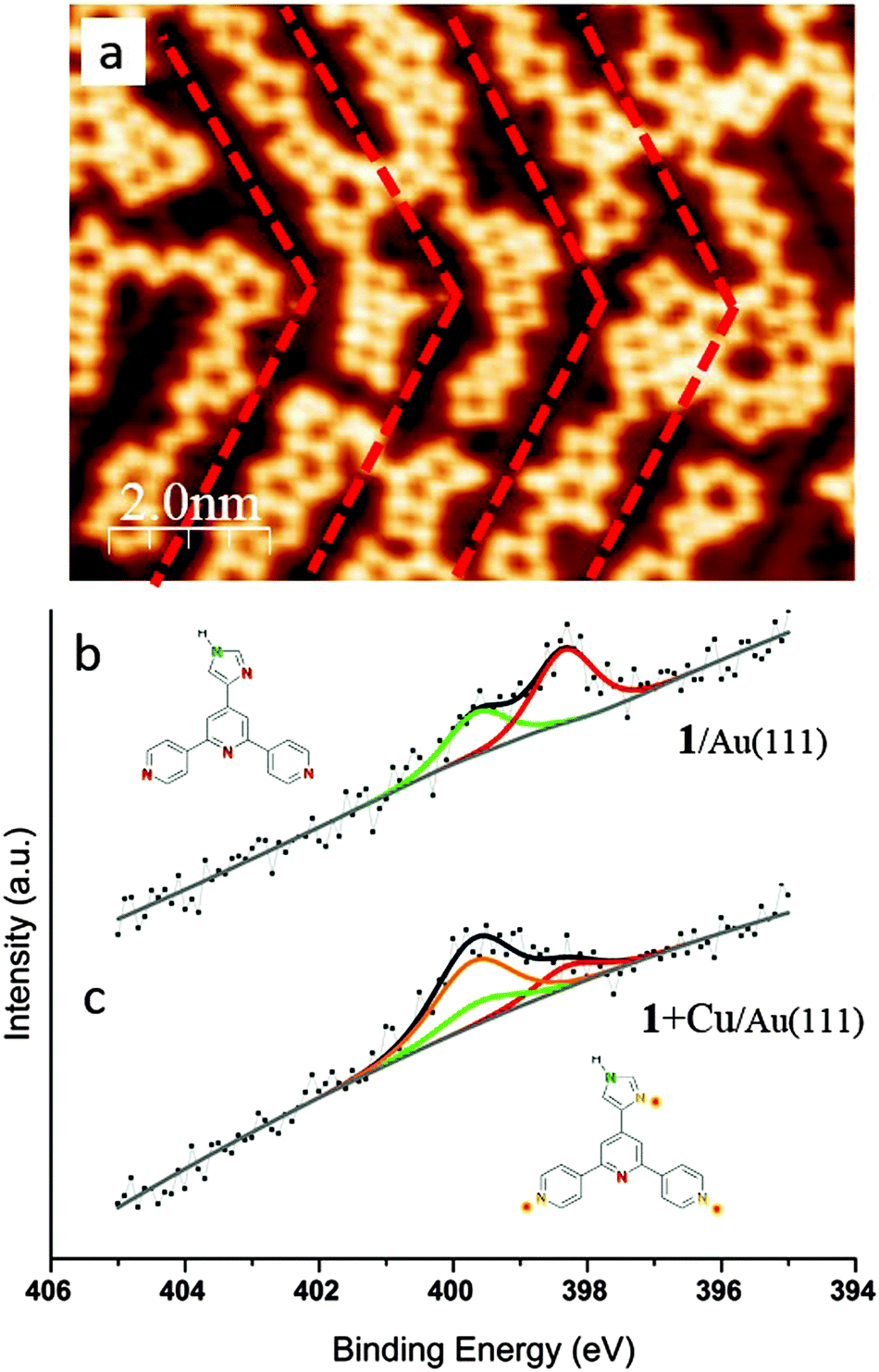 | ||
| Fig. 2 Metal coordination of 1 on Au(111). (a) STM image shows the metal coordinated structures, which are oriented along the fcc region of the Au(111) herringbone reconstruction (highlighted in red; see also Fig. S4†). (b and c) XPS spectra showing the N environment of 1 before (b) and after (c) deposition of copper adatoms. The peak deconvolution reveals two uncoordinated species (the central Ntpy and NHimidazole, red and green respectively), whereas the remaining N energy shift upwards consistent with coordination. | ||
The presence of various sized and shaped macrocycles (Fig. 3a) is consistent with (i) metal-binding through only the outer N-donors of the 4,2′:6′,4′′-tpy unit (as confirmed crystallographically for 4,2′:6′,4′′-tpy complexes10) and (ii) a balance between molecule–molecule and increased molecule–substrate interactions. The internal angle of the divergent 4,2′:6′,4′′-tpy domain is 120° and ideally matched to a hexameric assembly. However, the histogram in Fig. 3b reveals that this 6-fold assembly appears only in a minority of on-surface motifs which comprise 3-, 4-, 5- and 6-membered macrocycles, indicating the presence of an additional limiting factor beyond the macrocycle ring strain. This can be attributed to the width of the fcc region of the herringbone reconstruction, limiting the area of the macrocycles.23 The 6-membered macrocycle represents the most favourable angle configuration, the 4-membered the most favourable size configuration, and the presence of the 5-membered macrocycles represents a compromise of both cases. In each of the 4-, 5- and 6-membered metallomacrocycles, coordination involving only the 4,2′:6′,4′′-tpy domain is proposed. However, this coordination mode would lead to a strained 3-membered cyclic array. Comparison of the four images in Fig. 3a clearly reveals that the larger rings have 6-, 5- and 4-fold symmetry, whereas the trimer appears ‘squashed’ and is not 3-fold symmetric; furthermore, 3-membered rings are only observed as motifs on the periphery of larger rings. Metal binding involving both Ntpy and Nimidazole is consistent with these observations (Fig. 3a, right). Other examples are known, where the Au(111) herringbone reconstruction guides the molecular assembly. The fcc and hcp differ not only in topography, but also possess different electronic properties (the hcp stacked top layer is more electron rich).24 Therefore, molecules have preferential adsorption sites,25 especially if there is a strong interaction with the substrate (e.g. via dipolar interactions26 or via metal coordination4).
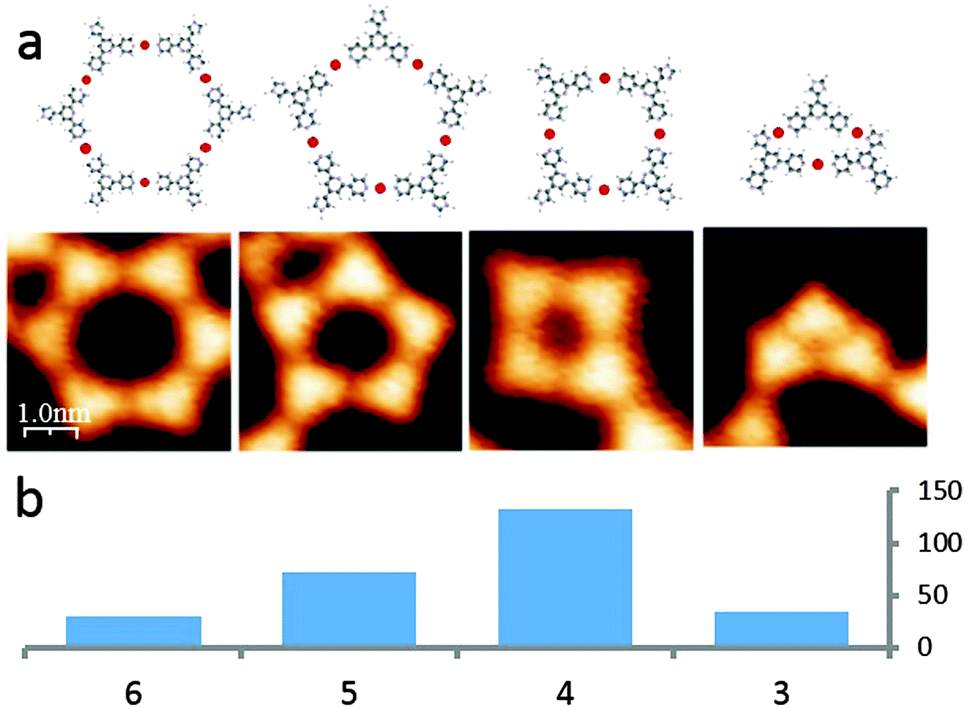 | ||
| Fig. 3 Macrocycle distribution. (a) Models and respective STM images of the most appearing macrocycles. (b) Histogram reveals existence of preferential pore-geometry, which are macrocycles consisting out of 3, 4, 5 or 6 molecular building blocks. | ||
In conclusion, we have shown that 1 assembles on a Au(111) surface into a close-packed phase which co-exists with a 6-fold nanoporous structure. The prochirality of 1 results in each hexacycle possessing a handedness and the chirality persists throughout the domain, with domains of both chiralities being present. Best-fit models are consistent with NHimidazole⋯Ntpy hydrogen bonds being the dominant interactions. The introduction of copper adatoms switches the on-surface assembly to discrete cyclic structures, the size distribution of which is consistent with the Au(111) herringbone reconstruction guiding the molecular assembly.
We thank the Swiss Nano Institute, Swiss National Science Foundation (Grant 200020_149067), São Paulo Research Foundation FAPESP (Grant 2013/04855-0) and the University of Basel for support. Marco Martina is thanked for supporting the infrastructure and co-developing and maintaining the sample preparation and handling procedures; a prototype of a vacuum suitcase (KTI PSPM and Ferrovac Inc) has been used in this work. Srboljub Vujovic is thanked for supplying ligand 1.
References
- C. A. Palma, M. Cecchini and P. Samori, Chem. Soc. Rev., 2012, 41, 3713 RSC; J. V. Barth, Surf. Sci., 2009, 603, 1533 CrossRef CAS PubMed.
- F. Bebensee, K. Svane, C. Bombis, F. Masini, S. Klyatskaya, F. Besenbacher, M. Ruben, B. Hammer and T. R. Linderoth, Angew. Chem., Int. Ed., 2014, 53, 12955 CrossRef CAS PubMed and references cited therein.
- M. Matena, J. Björk, M. Wahl, T.-L. Lee, J. Zegenhagen, L. H. Gade, T. A. Jung, M. Persson and M. Stöhr, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 235419 CrossRef.
- A. Shchyrba, M.-T. Nguyen, C. Wäckerlin, S. Martens, S. Nowakowska, T. Ivas, J. Roose, T. Nijs, S. Boz, M. Schär, M. Stöhr, C. A. Pignedoli, C. Thilgen, F. Diederich, D. Passerone and T. A. Jung, J. Am. Chem. Soc., 2013, 135, 15270 CrossRef CAS PubMed.
- S. L. Tait, Y. Wang, G. Costantini, N. Lin, A. Baraldi, F. Esch, L. Petaccia, S. Lizzit and K. Kern, J. Am. Chem. Soc., 2008, 130, 2108 CrossRef CAS PubMed.
- J. Reichert, M. Marschall, K. Seufert, D. Ecija, W. Auwärter, E. Arras, S. Klyatskaya, M. Ruben and J. V. Barth, J. Phys. Chem. C, 2013, 117, 12858 CAS.
- See for example: Y. Li, J. Xiao, T. E. Shubina, M. Chen, Z. Shi, M. Schmid, H.-P. Steinrück, J. M. Gottfried and N. Lin, J. Am. Chem. Soc., 2012, 134, 6401 CrossRef CAS PubMed; T. Lin, Q. Wu, J. Liu, Z. Shi, P. N. Liu and N. Lin, J. Chem. Phys., 2015, 142, 101909 CrossRef PubMed; G. Lyu, R. Zhang, X. Zhang, P. N. Liu and N. Lin, J. Mater. Chem. C, 2015, 3, 3252 RSC.
- See for example: J. I. Urgel, D. Ecija, W. Auwärter, D. Stassen, D. Bonifazi and J. V. Barth, Angew. Chem., Int. Ed., 2015, 54, 6163 CrossRef CAS PubMed; S. Vijayaraghavan, D. Ecija, W. Auwärter, S. Joshi, K. Seufert, M. Drach, D. Nieckarz, P. Szabelski, C. Aurisicchio, D. Bonifazi and J. V. Barth, Chem. – Eur. J., 2013, 19, 14143 CrossRef PubMed; D. Heim, D. Écija, K. Seufert, W. Auwärter, C. Aurisicchio, C. Fabbro, D. Bonifazi and J. V. Barth, J. Am. Chem. Soc., 2010, 132, 6783 CrossRef PubMed.
- E. C. Constable, Chem. Soc. Rev., 2007, 36, 246 RSC; E. C. Constable, Chimia, 2013, 67, 388 CrossRef CAS PubMed and references cited therein.
- C. E. Housecroft, Dalton Trans., 2014, 43, 6594 RSC.
- J. Wang and G. S. Hanan, Synlett, 2005, 1251 CAS.
- L. S. Pinheiro and M. L. A. Temperini, Surf. Sci., 2000, 464, 176 CrossRef CAS.
- See for example: P. R. Andres, R. Lunkwitz, G. R. Pabst, K. Böhn, D. Wouters, S. Schmatloch and U. S. Schubert, Eur. J. Org. Chem., 2003, 3769 CrossRef CAS PubMed; D. J. Díaz, S. Bernhard, G. D. Storrier and H. D. Abruña, J. Phys. Chem. B, 2001, 105, 8746 CrossRef; C. Grave, D. Lentz, A. Schäfer, P. Samori, J. P. Rabe, R. Franke and A. D. Schlüter, J. Am. Chem. Soc., 2003, 125, 6907 CrossRef PubMed; E. C. Constable, H.-J. Güntherodt, C. E. Housecroft, L. Merz, M. Neuburger, S. Schaffner and Y. Tao, New J. Chem., 2006, 20, 1470 RSC; T. Albrecht, K. Moth-Poulsen, J. B. Christensen, A. Guckian, T. Bjørnholm, J. G. Vos and J. Ulstrup, Faraday Discuss., 2006, 131, 265 RSC; E. Figgemeier, L. Merz, B. A. Hermann, Y. C. Zimmermann, C. E. Housecroft, H.-J. Guntherodt and E. C. Constable, J. Phys. Chem. B, 2003, 107, 1157 CrossRef; W. Wang, S. Wang, Y. Hong, B. Z. Tang and N. Lin, Chem. Commun., 2011, 47, 10073 RSC; X. Q. Shi, W. H. Wang, S. Y. Wang, N. Lin and M. A. Van Hove, Catal. Today, 2011, 177, 50 CrossRef PubMed; D. Trawny, P. Schlexer, K. Steenbergen, J. P. Rabe, B. Paulus and H.-U. Reissig, ChemPhysChem, 2015, 16, 949 CrossRef PubMed; S. L. Tait, A. Langner, N. Lin, S. Stepanow, C. Rajadural, M. Ruben and K. Kern, J. Phys. Chem. C, 2007, 111, 10982 Search PubMed.
- S. Wang, F. Zhao, S. Luo, Y. Geng, Q. Zeng and C. Wang, Phys. Chem. Chem. Phys., 2015, 17, 12350 RSC.
- E. C. Constable, C. E. Housecroft, M. Neuburger, S. Vujovic, J. A. Zampese and G. Zhang, CrystEngComm, 2012, 14, 3554 RSC.
- J. V. Barth, H. Brune, G. Ertl and R. J. Behm, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 9307 CrossRef CAS and references therein.
- Z. Shi and N. Lin, J. Am. Chem. Soc., 2009, 131, 5376 CrossRef CAS PubMed.
- F. Klappenberger, D. Kühne, W. Krenner, I. Silanes, A. Arnau, F. J. García de Abajo, S. Klyatskaya, M. Ruben and J. V. Barth, Nano Lett., 2009, 9, 3509 CrossRef CAS PubMed.
- J. S. Stevens, A. C. de Luca, M. Pelendritis, G. Terenghi, S. Downes and S. L. M. Schroeder, Surf. Interface Anal., 2013, 45, 1238 CrossRef CAS PubMed.
- J. M. L. Martínez, E. Rodríguez-Castellón, R. M. Torres Sánchez, L. R. Denaday, G. Y. Buldain and V. Campo Dall' Orto, J. Mol. Catal., 2011, 339, 43 CrossRef PubMed.
- A. Shchyrba, C. Wäckerlin, J. Nowakowski, S. Nowakowska, J. Björk, S. Fatayer, J. Girovsky, T. Nijs, S. C. Martens, A. Kleibert, M. Stöhr, N. Ballav, T. A. Jung and L. H. Gade, J. Am. Chem. Soc., 2014, 136, 9355 CrossRef CAS PubMed.
- C. H.-H. Traulsen, E. Darlatt, S. Richter, J. Poppenberg, S. Hoof, W. E. S. Unger and C. A. Schalley, Langmuir, 2012, 28, 10755 CrossRef CAS PubMed; D. Skomski, C. D. Tempas, K. A. Smith and S. L. Tait, J. Am. Chem. Soc., 2014, 136, 9862 CrossRef PubMed.
- L.-A. Fendt, M. Stöhr, N. Wintjes, M. Enache, T. A. Jung and F. Diederich, Chem. – Eur. J., 2009, 15, 11139 CrossRef CAS PubMed.
- W. Chen, V. Madhavan, T. Jamneala and M. Crommie, Phys. Rev. Lett., 1998, 80, 1469 CrossRef CAS.
- M. E. Cañas-Ventura, K. Aït-Mansour, P. Ruffieux, R. Rieger, K. Müllen, H. Brune and R. Fasel, ACS Nano, 2011, 5, 457 CrossRef CAS PubMed; Y. Pan, B. Yang, C. Hulot, S. Blechert, N. Nilius and H.-J. Freund, Phys. Chem. Chem. Phys., 2012, 14, 10987 RSC; I. Fernandez-Torrente, S. Monturet, K. J. Franke, J. Fraxedas, N. Lorente and J. I. Pascual, Phys. Rev. Lett., 2007, 99, 176103 CrossRef; M. Yu, N. Kalashnyk, R. Barattin, Y. Benjalal, M. Hliwa, X. Bouju, A. Gourdon, C. Joachim, E. Lægsgaard, F. Besenbacher and T. R. Linderoth, Chem. Commun., 2010, 46, 5545 RSC.
- M. Böhringer, K. Morgenstern, W.-D. Schneider, M. Wühn, C. Wöll and R. Berndt, Surf. Sci., 2000, 444, 199 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional Fig. S1–S5; Tables S1 and S2 with XPS data. See DOI: 10.1039/c5cc04186d |
| This journal is © The Royal Society of Chemistry 2015 |