
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum trimethyl bipyridyl thiolates – new, tunable, red- to near IR emitting luminophores for bioimaging applications†
Harriet L.
Steel
a,
Sarah L.
Allinson
a,
Jane
Andre
a,
Michael P.
Coogan
*b and
James A.
Platts
c
aDivision of Biomedical and Life Sciences, Faculty of Health and Medicine, Furness Building, Lancaster University, Bailrigg, Lancaster, LA1 4YG, UK
bChemistry Department, Faraday Building, Lancaster University, Bailrigg, Lancaster, LA1 4YB, UK. E-mail: m.coogan@lancaster.ac.uk
cCardiff School of Chemistry, Cardiff University, Park Place, Cardiff, CF10 3AT, UK
First published on 16th June 2015
Abstract
Synthetic, spectroscopic, computational and biological imaging studies of platinum trimethyl bipyridyl thiolate complexes of the general formula [PtMe3(bpy)SR] reveal these to be easily accessed, tunable bioimaging agents which feature an unusual σ–π* Inter-Ligand Charge Transfer (ILCT) transition, and in some cases emit into the Near infra-red (NIR).
Platinum(IV) trimethyl iodide exists as a cubic tetramer which reacts with a large range of ligands to give stable octahedral complexes of the general formula fac-[Pt(Me)3(L)2I] which have been extensively studied.1 In the case of chelating bis-heterocyclic ligands such as bipyridine, the derived complexes, e.g. fac-[Pt(Me)3(N^N)I] 1, are luminescent, showing room temperature phosphorescence from excited states assigned as 3IL π–π*.1 The photochemistry of these units has been widely explored, with photo-reduction to the square-planar Pt(II) species typically observed,2 but there are few applications of Pt(IV)Me3 complexes in luminescence, while there is much data on cyclometallated Pt(II) complexes as lumophores.3–5
The analogous fac-[Re(CO)3(N^N)L] complexes are widely applied lumophores6 in which systematic variations in the (N^N) unit tune the absorption and emission characteristics, while variations in L (usually substituted pyridines) control solubility and other physical characteristics.7 It is generally the case that substituted complexes, in which L is a nitrogen heterocycle, have attractive photophysical properties and stability while the precursor halido-complexes are unstable and tend to be toxic in biological work due to halide lability.8 Therefore, an investigation was undertaken of the synthesis and photophysical characteristics of a series of fac-Pt(Me)3 complexes of chelating and monodentate ligands. Reaction of PtIMe3 with substituted bipyridines gave analogues of 1 with electron donating (OMe, complex 2) and withdrawing (CO2Me, complex 3) groups in the 4,4′ positions (Scheme 1).
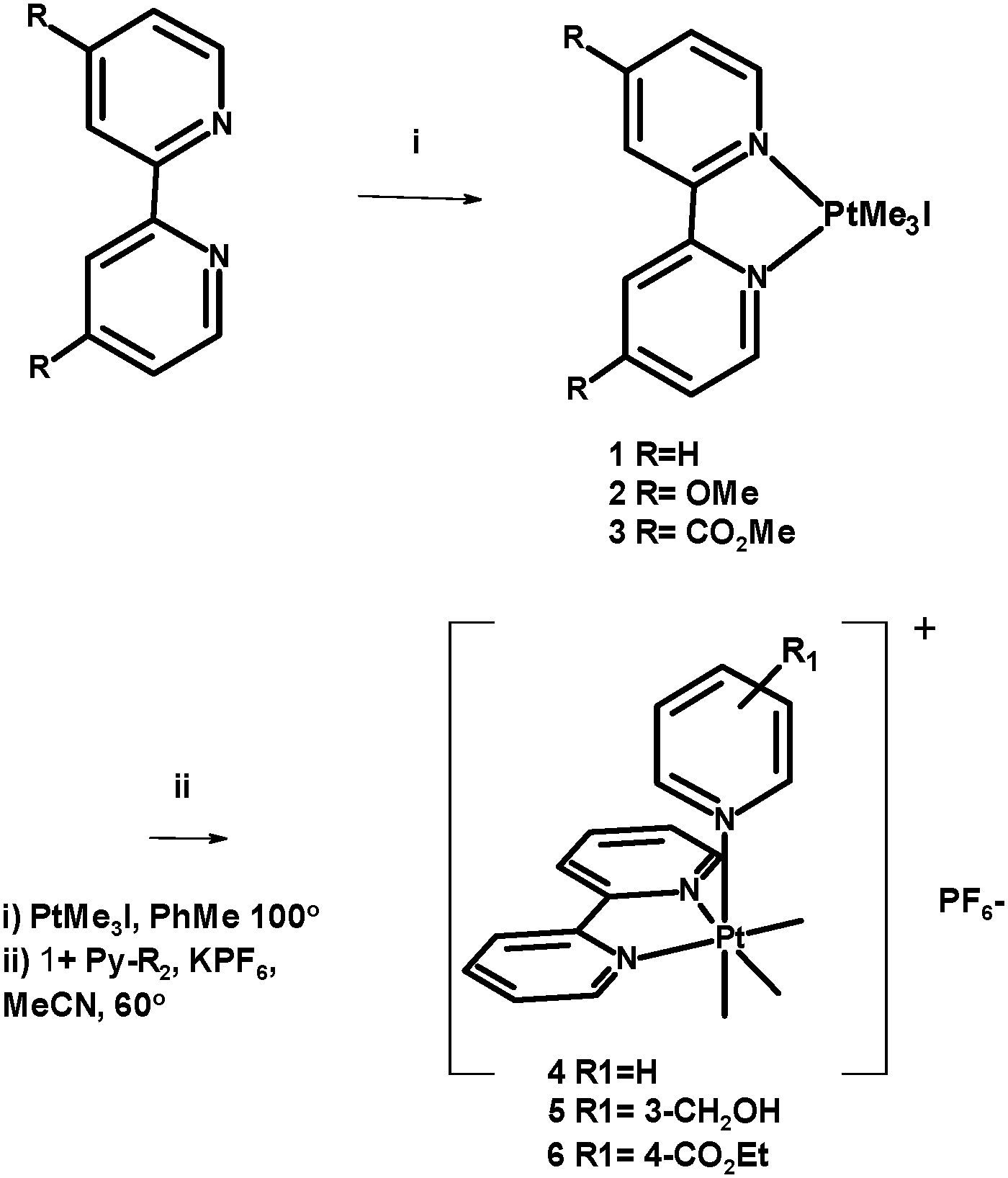 | ||
| Scheme 1 Synthesis of complexes 1–6. | ||
These complexes showed the respective blue- and red-shifted absorptions and emissions which would be predicted from the electronic structures (Table 1). Complexes of the general formula fac-[Pt(Me)3(bpy)L], where L is a substituted pyridine, were easily synthesised by the reaction of 1 with the appropriate pyridine in the presence of KPF6 to give complexes 4–6 (Scheme 1). However, the yellow colour of 1 had been lost, and while these complexes were luminescent (see Table 1) UV excitation was required. Reaction of 1 with triphenyl phosphine or aniline gave complexes 7 and 8, but these complexes too required u.v. excitation. As it seemed that substituting the iodide led to loss of the low energy band responsible for visible absorption and excitation, a re-examination of the nature of this band was required. It is not clear why substitution of iodide for N- and P-donor ligands leads to complete loss of this transition, which has previously been assigned as a formally disallowed 3IL π–π* transition.1
| l max absa | l max absb | l max ex | l max em | Quantum yield Φc | |
|---|---|---|---|---|---|
| a Strongest bands >275 nm. b Lowest energy band/shoulder. c Measurements performed irradiating into ILCT band for each complex in aerated acetonitrile solutions, using [Ru(bpy)3](PF6)2 as a standard (Φem = 0.018)17. d ILCT band absent. | |||||
| 1 | 297, 308 | 344 | 380 | 530 | See ref. 1 |
| 2 | 280, 289, 299 | 356 | 330 | 520 | 1.3 × 10−2 |
| 3 | 314, 323 | 372 | 420 | 600 | 2.6 × 10−4 |
| 4 | 300, 311 | — | 330 | 470 | |
| 5 | 300, 310 | — | 333 | 470 | |
| 6 | 290, 300, 310 | — | 330 | 470 | |
| 7 | 304, 313 | — | 325 | 470 | |
| 8 | 290, 298, 310 | — | 345 | 540 | |
| 9 | 301, 308, 358 | 440 | 450 | 660 | 4.1 × 10−4 |
| 10 | 310, 350 | 500 | 490 | 560 | 3.7 × 10−5 |
We therefore carried out time-dependent density functional theory (TD-DFT)9–16 calculations on 1 and 4 to compare the absorption bands in these species in detail (see ESI† for details). This predicts absorption bands for 1 at 358 and 294 nm, in reasonable agreement with the experimental values of 344, 308 and 297 nm. In contrast, 4 is predicted to absorb at 294 and 284 nm (experimental values 311 and 300 nm). The band at 358 nm in 1 corresponds to excitation from an orbital made up largely of a Pt–I σ-bond, along with a lesser amount of trans-Pt–Me, to the π* MO on bpy, as illustrated in Fig. 1. Hence this low energy band is best described as Inter-Ligand Charge Transfer rather than either 3IL π–π* or MLCT. ILCT between halogen and bipyridine antibonding orbitals is also seen admixed with MLCT in the analogous Re complexes,6 which occasionally may cause problems in biological studies due to halide lability leading to interaction of the heavy metal centre with biomolecules (e.g. DNA).18 These results therefore call into question the literature assignment, but do explain the change in absorption patterns between 1 and 4, since the latter does not contain a low energy orbital of the correct symmetry to reproduce such absorption. They also suggest a strategy for combining the desirable properties of visible absorption and kinetic stability in [Pt(Me3)N^N L]+ complexes, i.e. to find L that combines a strong Pt–L bond with suitable electronic structure for interaction with bpy π*. Thiols satisfy both requirements, so further TD-DFT calculations were performed for [Pt(Me3)(bpy)(SH)], from which we predict an absorption band at 393 nm corresponding to S lone pair/Pt–S σ-bond to bpy π* orbitals (Fig. 1).
 | ||
| Fig. 1 Occupied (bottom) and unoccupied (top) orbitals of 1, 4, Pt(Me)3(bipy)SH and 9 involved in low energy absorption bands. | ||
Having obtained computational support for the hypothesis that thio-substituted complexes should show low energy absorption, methyl-4-mercaptobenzoate was selected as a model ligand which would (through variations of the ester) allow the incorporation of a variety of substituents at the sulphur ligand, giving the tuneable lipophilicity etc. which has proven important in the development of metallo-imaging agents. Treatment of 1 with a small excess of methyl-4-mercaptobenzoate in acetonitrile in the presence of sodium t-butoxide gave the thiobenzoate-substituted complex 9 (Scheme 2: full data in ESI†), and reaction with 3 gave the ester-substituted analogue 10. Electronic spectroscopy confirmed that 9 exhibits low energy absorption, observed as a shoulder centred at 440 nm, along with higher energy bands around 300/350 nm (TD-DFT predicts absorption at 440 nm due to S LP → π*/S–Pt → π*, Fig. 1). Luminescence spectroscopy indicated that the maximum excitation band was centred at ca. 450 nm, correlating with the likely true maximum of the band observed as a shoulder at 440 nm in the UV-vis spectrum. Exciting at 450 nm gave rise to intense emission as a broad band centred around 660 nm (Fig. 2).
 | ||
| Scheme 2 Synthesis of complexes 9 and 10. (i) MeCN, NaO-t-Bu. | ||
 | ||
| Fig. 2 Excitation and emission spectra for 9 and 10. | ||
Complex 9 is air- and water stable, resistant to ligand substitution of the coordinated thiolate under physiological conditions, and with visible excitation, a Stokes shift of over 200 nm and red emission is an ideal candidate for fluorescence imaging experiments.
Complex 3 bearing electron withdrawing ester substituents was likewise converted to the methyl 4-mercaptobenzoate complex 10 and showed a low energy absorption band centred at ca. 500 nm (DFT prediction 532 nm), and an excitation maximum at 490 nm, (Fig. 2) confirming that the photophysics of these complexes is susceptible to significant variations accessible by ligand variations. However, unexpectedly the emission maximum of 10 was blue-shifted in comparison to that of 9 indicating that simple assumptions regarding substituent effects can be misleading, especially in the case of emission from triplet states where the Stokes shift is a function of energy losses through relaxation into triplet geometries, the magnitude of which cannot be estimated intuitively from electron donating/withdrawing arguments.
As a preliminary assessment of the potential for applications of complexes such as 9, a study of cellular uptake was undertaken by flow cytometry and fluorescence microscopy. This used the well characterised HeLa human cervical carcinoma cell line, and was performed at 0–4 °C to inhibit endocytosis. Flow cytometry detecting between 655–735 nm (Fig. 3, PerCP-Cy5.5) appeared to indicate little increase in fluorescence over background at <125 μg mL−1, then a dramatic increase in uptake due to interference from autofluorescence at lower concentrations. However, taking advantage of the emission band of 9 which extends well into the NIR, detection between 750 and 810 nm (Fig. 3, PerCP-Cy7) clearly showed good uptake of 9 with a dose-dependent response with the number of cells showing enhanced intensity of emission increasing as a function of concentration.
 | ||
| Fig. 3 Flow cytometric analysis of 9 uptake by HeLa cells; x-axis = fluorescence intensity, y-axis = counts. Flow cytometry histograms of gated cells treated with the indicated concentrations of 9 for 10 minutes are shown. The PerCP-Cy5.5 channel detects fluorescence between 655 and 735 nm and the PE-Cy7 channel detects fluorescence between 750 and 810 nm. | ||
Confocal microscopy of HeLa S3 cells confirmed the dose-dependent cellular uptake observed in the flow cytometry correlated with uptake in intact cells (Fig. 4). The luminescence from 9 formed a punctate pattern of discrete staining of compartments in the perinuclear region of the cytoplasm at 25 μg mL−1, and at 125 μg mL−1 more generalised staining of the cytoplasmic region was observed. 9 was not designed with any of the features associated with a preference for localisation in a particular organelle, but is a prototype of this new class of lumophores, so it was pleasing that it appeared not to be retained in the plasma membrane but is capable of permeating the cytoplasm, which bodes well for the design of targeted imaging agents. While some deformations of the membrane surfaces were observed in confocal microscopy even at 25 μg, flow cytometry forward scatter and side scatter profiles indicate cells remained largely intact, even at 125 μg mL−1 (see ESI†). The combination of NIR detection and the ability to penetrate the cell membrane without causing cell lysis is an indication that these complexes have promise as lumophores in cell imaging. While Pt(II) complexes have previously been applied in cellular imaging,19–21 and have a well-established role in therapy,22 we believe this to be the first application of Pt(IV) species in fluorescence microscopy.23 The possibility of theranostic approaches with a Pt(IV) imaging agent of low cytotoxicity, which could be tailored through imaging studies to target organisms and organelles of interest, and reduced, or photoactivated, to a highly cytotoxic Pt(II) species is particularly interesting.
 | ||
| Fig. 4 Microscopy images of HeLa S3 cells treated with 9 at the indicated concentrations. Images show overlay of brightfield and confocal fluorescence (excitation 458 nm 30 mW Ar laser 50% power output 16% transmission; emission longpass 560 nm filter) fields. | ||
In summary, complexes of the general structure [PtMe3(N^N)SR], where (N^N) represents a bisimine ligand such as 2,2′-bipyridine, are visible absorbing, red or NIR emitting lumophores which are easily synthesised in a few steps from commercially available materials. DFT data indicate that absorption and excitation stems from promotion of an electron localised in the S–Pt bond into the π* orbital on bipyridine. Absorption and emission profiles are therefore tuneable through simple ligand substitutions. Preliminary experiments show that biological applications as fluorescent agents are possible with these complexes. The NIR emission can be used to differentiate agent-based emission from autofluorescence even at low levels of uptake and emission intensity.
We thank the BBSRC and Boots UK Ltd for funding (HLS), the EPSRC mass spectrometry service, Swansea for mass spectra, Dr Fraser White, Daniel Baker and Marcus Winter (Agilent Technologies, Yarnton, UK) for assistance with the X-ray structures of 2–4, the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data for 9 and 1024 and Prof. Peter Heard (Sunway University) for drawing our attention to the analogy between the [PtMe3] and [Re(CO)3] cations.
Notes and references
- H. Kunkely and A. Vogler, Coord. Chem. Rev., 1991, 111, 15 CrossRef CAS.
- D. C. L. Perkins, R. J. Puddephatt and C. F. H. Tipper, J. Organomet. Chem., 1979, 166, 261 CrossRef CAS.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762 CrossRef CAS PubMed.
- S.-W. Lai and C.-M. Che, Top. Curr. Chem., 2004, 241, 27 CAS.
- I. Eryazici, C. N. Moorefield and G. R. Newkome, Chem. Rev., 2008, 108, 1834 CrossRef CAS PubMed.
- A. Coleman, C. Brennan, J. G. Vos and M. T. Pryce, Coord. Chem. Rev., 2008, 252, 2585 CrossRef CAS PubMed; M. Bartholomä, J. Valliant, K. P. Maresca, J. Babich and J. Zubieta, Chem. Commun., 2009, 493 RSC; S. Clede and C. Policar, Chem. – Eur. J., 2015, 21, 942 CrossRef PubMed.
- L. A. Worl, R. Duesing, P. Chen, L. D. Ciana and T. J. Meyer, Dalton Trans., 1991, 849 RSC.
- R. G. Balasingham, M. P. Coogan and F. L. Thorp-Greenwood, Dalton Trans., 2011, 41, 11663 RSC.
- M. J. Frisch, et al., Gaussian 09, Revision C. 01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chem. Acc., 1990, 77, 123–141 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS PubMed.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed , and references cited therein.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishic and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850 RSC.
- A. J. Amoroso, M. P. Coogan, J. E. Dunne, V. Fernández-Moreira, J. B. Hess, A. J. Hayes, D. Lloyd, C. Millet, S. J. A. Pope and C. Williams, Chem. Commun., 2007, 3066 RSC.
- C. Yik-Sham Chung, S. Po-Yam Li, M.-W. Louie, K. K.-W. Lo and V. Wing-Wah Yam, Chem. Sci., 2013, 4, 2453 RSC.
- S. W. Botchway, M. Charnley, J. W. Haycock, A. W. Parker, D. L. Rochester, J. A. Weinstein and J. A. G. Williams, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16071 CrossRef CAS PubMed.
- R. R. de Haas, R. P. M. van Gijlswijk, E. B. van der Tol, J. Veuskens, H. E. van Gijssel, R. B. Tijdens, J. Bonnet, N. P. Verwoerd and H. J. Tanke, J. Histochem. Cytochem., 1999, 47, 183 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573 CrossRef CAS PubMed.
- M. P. Coogan and V. Fernández-Moreira, Chem. Commun., 2014, 50, 384 RSC.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and spectroscopic and crystallographic details of new materials, further details of DFT calculations, predicted absorption/emission spectra and orbitals involved. CCDC 1045315–1045320. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04003e |
| This journal is © The Royal Society of Chemistry 2015 |