
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic meta-selective C–H functionalization to construct quaternary carbon centres†
Andrew J.
Paterson
a,
Sahra
St John-Campbell
b,
Mary F.
Mahon
b,
Neil J.
Press
c and
Christopher G.
Frost
*ab
aCentre for Sustainable Chemical Technologies, University of Bath, Bath, BA2 7AY, UK
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: c.g.frost@bath.ac.uk; Fax: +44 (0)1225 386231; Tel: +44 (0)1225 386142
cNovartis Institutes for BioMedical Research, Novartis Campus, Fabrikstrasse, 22, CH-4056, Basel, Switzerland
First published on 6th July 2015
Abstract
A catalytic meta-selective C–H functionalization of 2-phenylpyridines using a range of tertiary halides is described. The protocol is simple to perform and uses commercially available reagents to construct challenging quaternary carbon centres in a regioselective manner. Preliminary studies suggest the C–H functionalization proceeds through a radical process directed via a remote σ-activation.
The transition-metal catalyzed cleavage and functionalization of inert C–H bonds is evolving into a fundamental methodology for the design of atom economical approaches to useful organic molecules.1 While the direct functionalization at the ortho position of aromatic compounds by chelation assisted C–H bond cleavage has become well established in recent years, developing reactions with complementary regioselectivity continues to challenge contemporary catalytic methodology.2 In this context, examples of meta selective catalytic C–H functionalization have been reported offering diversity in molecular design through alternative reaction strategies (Scheme 1a). These include substrate controlled systems,3 transient mediators such as a carboxylic acid4 or norbornene5 and covalent template strategies for remote activation.6 We first reported a novel catalytic σ-activation protocol for C–H functionalization that allows the meta sulfonation of 2-phenylpyridines via cyclometalated ruthenium intermediates.7 Interestingly, the catalytic σ-activation strategy proved effective for meta-alkylations with secondary alkyl halides8 whilst acyl halides and primary alkyl halides afford only the ortho-functionalized products consistent with a mechanism involving oxidative addition of the organohalide.9
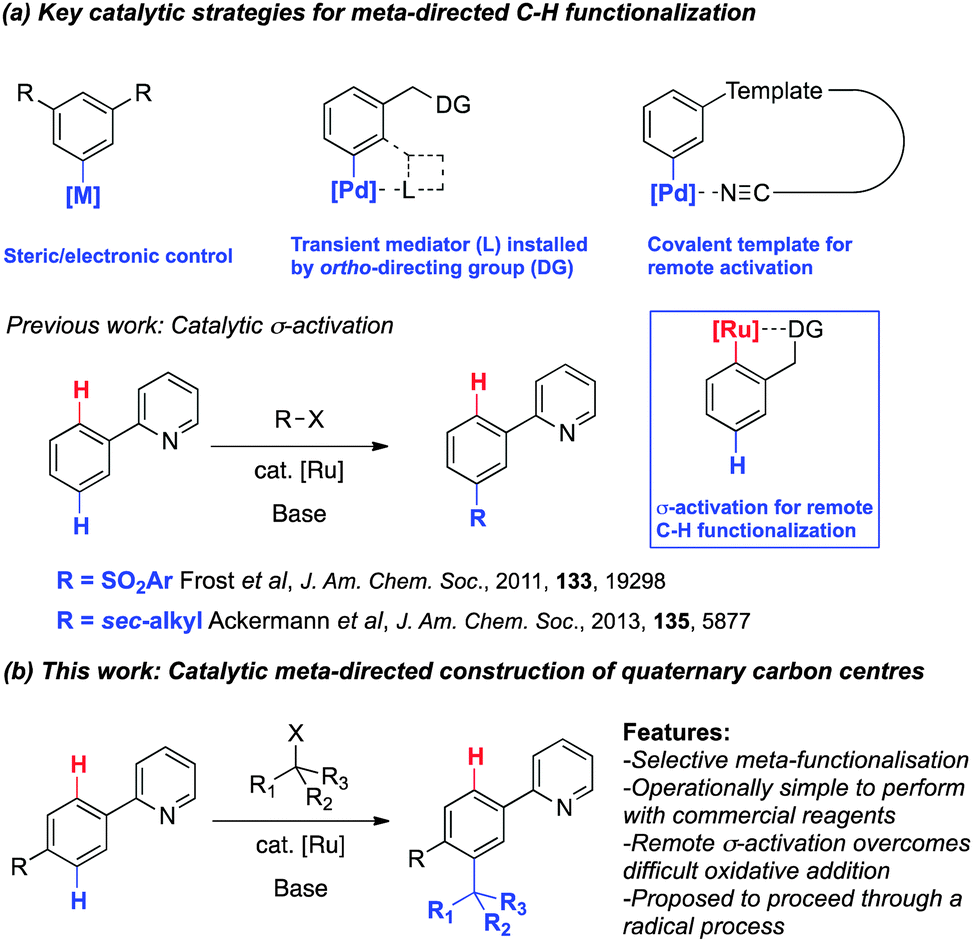 | ||
| Scheme 1 Catalytic meta-directed C–H functionalization. | ||
Here we report a new catalytic meta-selective C–H functionalization of 2-phenylpyridines to construct quaternary carbon centres (Scheme 1b). The transition-metal catalyzed coupling of tertiary alkyl halides and aromatic C–H bonds is an especially challenging reaction due to the difficult oxidative addition of a metal complex into a bulky C–X bond.10 We hypothesized that a catalytic σ-activation strategy would therefore be amenable to establishing quaternary carbon centres by avoiding a general oxidative addition pathway.
In preliminary experiments, 2-phenylpyridine 1a was treated under conditions analogous to those developed in our meta-sulfonation reaction: [RuCl2(p-cymene)]2 (5 mol%) K2CO3 (2 equiv.), t-BuBr 2a (3 equiv.) using MeCN as the solvent.7 Unfortunately no coupled products were formed under these conditions however the desired meta-substituted product was observed in 12% conversion when the reaction solvent was changed to 1,4-dioxane (Table 1, entries 1 and 2). By simply changing the base from K2CO3 to various acetate salts, a significant increase in conversion was observed with KOAc proving the most effective (entry 6). In the absence of ruthenium complex, no product was observed (entry 11). This catalytic system was found to perform well in a range of solvents as well as under solvent free conditions and was completed in as little as 4 hours (entry 14). When t-BuCl 2b was used as the coupling reagent, a significant drop in conversion was observed, however by using a combination of K2CO3 and KOAc, the reaction performed competitively (entry 17).
|
|
||||
|---|---|---|---|---|
| Entry | t-Bu-X | Base | Solvent | Conversiona (%) |
| a Conversion of 1a to 3a by 1H NMR. b With 30 mol% MesCOOH. c Without [RuCl2(p-cymene)]2. d Reaction in air. e [RuCl2(p-cymene)]2 (1 mol%). f Reaction time 4 h. | ||||
| 1 | 2a | K2CO3 | MeCN | 0 |
| 2 | 2a | K2CO3 | 1,4-Dioxane | 12 |
| 3 | 2a | KOAc | Neat | 69 |
| 4 | 2a | KOAc | 2-Me-THF | 68 |
| 5 | 2a | KOAc | 2-Butanone | 61 |
| 6 | 2a | KOAc | 1,4-Dioxane | 74 |
| 7b | 2a | K2CO3 | 1,4-Dioxane | 60 |
| 8 | 2a | NaOAc | 1,4-Dioxane | 31 |
| 9 | 2a | CsOAc | 1,4-Dioxane | 64 |
| 10 | 2a | Bu4NOAc | 1,4-Dioxane | 13 |
| 11c | 2a | KOAc | 1,4-Dioxane | 0 |
| 12d | 2a | KOAc | 1,4-Dioxane | 25 |
| 13e | 2a | KOAc | 1,4-Dioxane | 50 |
| 14f | 2a | KOAc | 1,4-Dioxane | 72 |
| 15 | 2b | KOAc | 1,4-Dioxane | 20 |
| 16 | 2b | K2CO3 | 1,4-Dioxane | 27 |
| 17 | 2b | KOAc (0.5 equiv.) K2CO3 (1.5 equiv.) | 1,4-Dioxane | 63 |
| 18b | 2b | K2CO3 | 1,4-Dioxane | 62 |
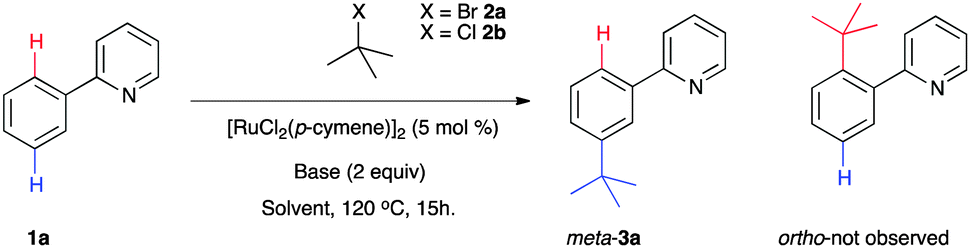
With optimized catalytic systems in hand, we then investigated how reaction conversions were affected when substituents at the 4-position of the aryl ring were varied (Scheme 2). It was found that electron donating substituents favoured the reaction whereas strongly electron withdrawing groups shut the reaction down entirely. The reaction was tolerant of halogen and ester substituents which is useful for further synthetic transformations. The reactions led to the sole formation of the mono substituted meta products with no decomposition or by-products observed although quantitative separation by conventional methods was not always possible (see ESI† for full analysis). Intriguingly, 1-bromoadamantane was found to be an effective coupling partner and product 4c was characterised by X-ray analysis confirming the regioselective meta substitution (Fig. 1).11 Our procedure also effectively coupled a range of tertiary alkyl chlorides, reagents which are readily available and generally considered to be less reactive (Scheme 3). In these examples, it was found that the incorporation of longer alkyl chain lengths maintained high conversions and enabled better separation of the products by normal phase flash chromatography.
 | ||
| Scheme 2 Catalytic meta functionalization using tertiary alkyl bromides. Numbers quoted are direct conversions to product by 1H NMR. a Using KOAc (0.5 equiv.) and K2CO3 (1.5 equiv.). | ||
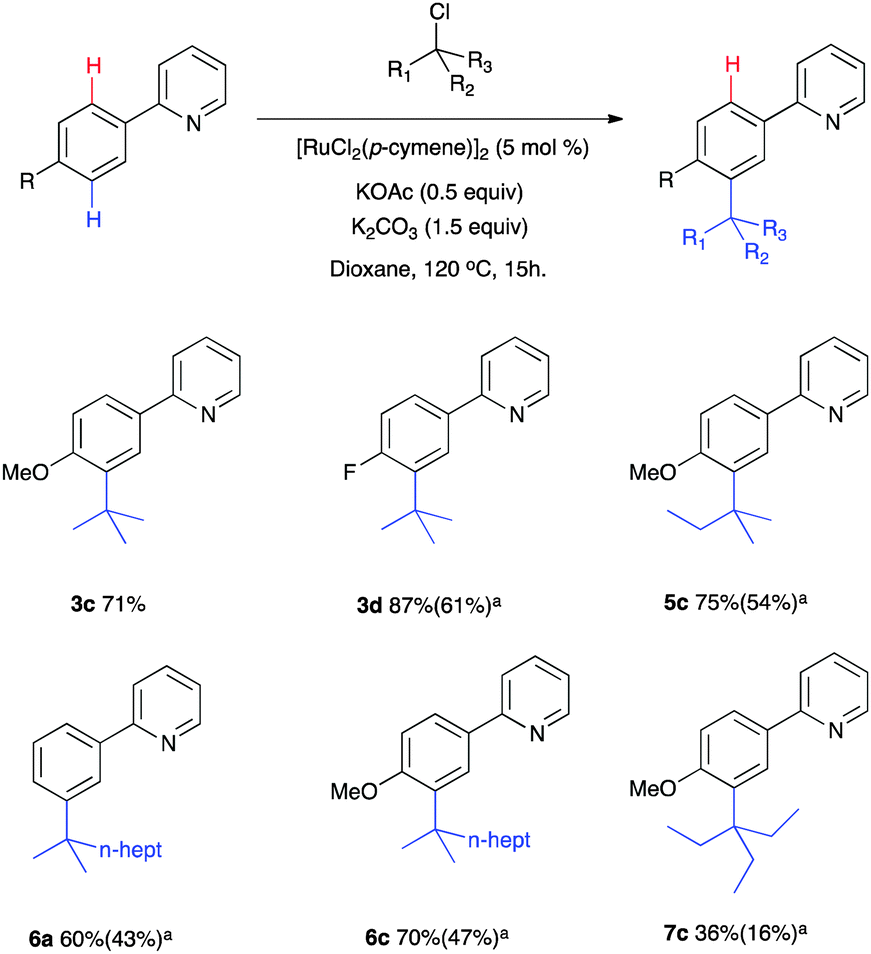 | ||
| Scheme 3 Catalytic meta functionalization using alkyl chloride reagents. Numbers quoted are direct conversions to product by 1H NMR. a Numbers in brackets indicate isolated yields. | ||
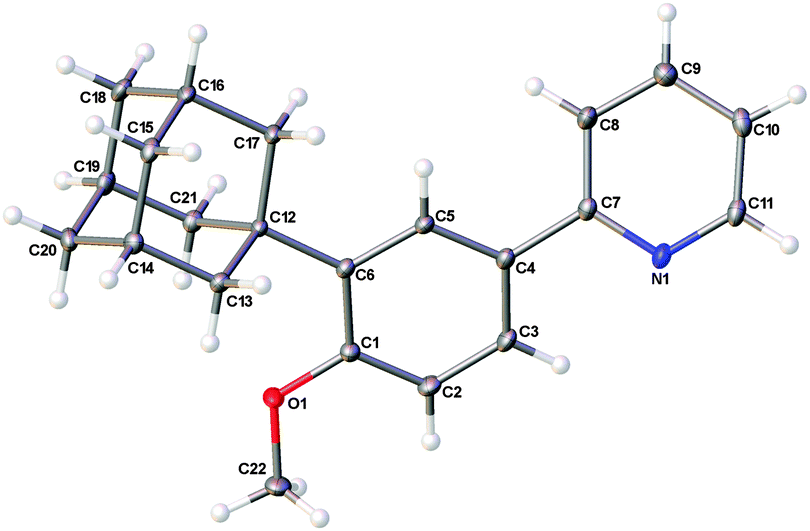 | ||
| Fig. 1 The asymmetric unit in the crystal structure of 4c. Ellipsoids are illustrated at 30% probability. | ||
In addition to the alkyl halide reagents outlined in Schemes 2 and 3, tertiary α-bromo ester 2c was effectively coupled, generating meta-substituted products 8a, 8c and 8d, compounds with a useful functional handle, in reasonable isolated yields (Scheme 4). This result provided key insight into the reaction mechanism and strongly suggested a radical type pathway, rather an SEAr type mechanism previously proposed in our meta-sulfonation reaction.7
 | ||
| Scheme 4 Catalytic meta functionalization with α-bromo ester 2c. Numbers quoted are isolated yields. | ||
Heterolytic cleavage of the C–X bond of 2c in an SN1-type manner would result in a strongly disfavoured carbocation residing alpha to an electron withdrawing ester. It is therefore unlikely that reaction with the aromatic substrate would occur in this fashion. The possibility of SN2 type reactivity can also be effectively ruled out given the steric effects of the tertiary alkyl halides used. The generation of tertiary alkyl radicals has however been widely reported with a range of transition metal catalysts and shown to be effective in the substitution of aromatics, heteroaromatics and olefins.12
In contrast to the reactions with simple alkyl-halides outlined in Schemes 2 and 3 which led to the sole formation of one product, reaction with 2c generated additional by-products. Compounds 9a, 9c and 9d were isolated along with spectroscopic evidence of trace higher oligomers which is consistent with a radical conjugate polymerisation pathway. We hypothesise that a tertiary carbon-centered radical species can add onto elimination products formed under the reaction conditions, which can in turn propagate onto a cyclometalated (σ-activated) substrate molecule to afford the observed by-products. Furthermore, the addition of radical scavenger TEMPO proved detrimental to the reaction with no desired product observed when stoichiometric quantities were used (see ESI†).
Further mechanistic work was conducted to provide additional insight into the interesting meta selectivity displayed by this reaction (Scheme 5). It has previously been proposed that initial ruthenium insertion into an ortho C–H bond to generate a cyclometalated complex is key to this type of reactivity.7,8 In support of this, reaction of the ortho, ortho dimethyl substrate 1i resulted in no conversion to the desired meta substituted product. The importance of ruthenium σ-activation is also highlighted with the successful meta-selective reaction using pre-formed complex A. No meta-substituted product was observed when substrate 1j bearing a methyl group at the 3-position of the aromatic ring was used. Instead, the only product isolated was dimer 10 suggesting a competing reductive elimination of two coordinated substrate molecules when the site para to the C–Ru bond is blocked.13 Conformationally locked benzoquinoline 1j was however effectively alkylated generating 10 as the only isolated product.
 | ||
| Scheme 5 Mechanistic investigation. Numbers quoted are isolated yields. | ||
Together these results suggest that substitution occurs preferentially at a position para to the C–Ru bond formed following cyclometalation. Interestingly, analogous reactivity has also recently been reported in a stoichiometric process on iridium complexes.14 In light of this work we now propose the following mechanism (Scheme 6). Initial ortho C–H insertion generates a cyclometalated complex, a process shown to be reversible and aided by carboxylate ligands.15 Substitution at the position para to the newly installed C–Ru bond then most likely occurs via a radical process whereby single-electron transfer (SET) from a ruthenium(II) species can generate a tertiary alkyl radical and the corresponding ruthenium(III)X species. The carbon-centered radical then adds to the aromatic ring to generate a cyclohexadienyl radical intermediate. Rearomatisation could occur via single-electron oxidation and deprotonation to regenerate a ruthenium(II) complex and furnish the meta alkylated product after proto-demetalation.
 | ||
| Scheme 6 Proposed catalytic cycle. | ||
In summary, we have developed a novel meta selective catalytic C–H functionalisation of 2-phenylpyridine substrates for the installation of quaternary carbon centres. The procedure is operationally simple and was found to couple a useful range of tertiary alkyl bromides and more challenging tertiary alkyl chlorides. Mechanistic studies indicate that site selective radical addition occurs at the position para to the C–Ru bond formed following cyclometalation to afford products with net meta substitution. More detailed mechanistic studies are underway to determine the precise nature of the organometallic species and redox processes involved.
We are grateful to the University of Bath, EPSRC DTC in Sustainable Chemical Technologies and Novartis for funding. We acknowledge the valuable assistance of Dr Anneke Lubben (Mass Spectrometry, University of Bath), Dr John Lowe and Dr Catherine Lyall (NMR Spectroscopy, University of Bath).
Notes and references
- For selected reviews, see: (a) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (b) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (c) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (d) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (e) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (f) Topics in Current Chemistry: C–H Activation, ed. J.-Q. Yu and Z. Shi, Springer, Berlin Heidelberg, 1st edn, 2010 Search PubMed.
- (a) F. Julia-Hernandez, M. Simonetti and I. Larrosa, Angew. Chem., Int. Ed., 2013, 52, 11458 CrossRef CAS PubMed; (b) J. Yang, Org. Biomol. Chem., 2015, 13, 1930 RSC.
- (a) H. A. Duong, R. E. Gilligan, M. L. Cooke, R. J. Phipps and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 463 CrossRef CAS PubMed; (b) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593 CrossRef CAS PubMed . For an excellent review of of non-chelate-assisted C–H activation, see: ; (c) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed.
- (a) J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109 CrossRef CAS PubMed; (b) S. Mochida, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 5776 CrossRef CAS PubMed; (c) J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429 CrossRef CAS PubMed.
- (a) X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334 CrossRef CAS PubMed; (b) Z. Dong, J. Wang and G. Dong, J. Am. Chem. Soc., 2015, 137, 5887 CrossRef CAS PubMed.
- For selected examples, see: (a) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518 CrossRef CAS PubMed; (b) H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567 CrossRef CAS PubMed; (c) L. Wan, N. Dastbaravardeh, G. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 18056 CrossRef CAS PubMed; (d) G. Yang, P. Lindovska, D. Zhu, J. Kim, P. Wang, R.-Y. Tang, M. Movassaghi and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 10807 CrossRef CAS PubMed; (e) R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215 CrossRef CAS PubMed.
- (a) O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298 CrossRef CAS PubMed; (b) W. R. Reynolds, P. M. Liu, G. Kociok-Köhn and C. G. Frost, Synlett, 2013, 2687 CAS.
- N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877 CrossRef CAS PubMed.
- (a) P. M. Liu and C. G. Frost, Org. Lett., 2013, 15, 5862 CrossRef CAS PubMed; (b) L. Ackermann, N. Hofmann and R. Vicente, Org. Lett., 2011, 13, 1875 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2001, 40, 4591 CrossRef CAS; (b) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2007, 5969 CrossRef CAS PubMed; (c) O. Riant and J. Hannedouche, Org. Biomol. Chem., 2007, 5, 873 RSC; (d) Y. Tu and B. Wang, Acc. Chem. Res., 2011, 44, 1207 CrossRef PubMed; (e) A. Y. Hong and B. M. Stoltz, Eur. J. Org. Chem., 2013, 2745 CrossRef CAS PubMed; (f) T. Nishikata and S. Ishikawa, Synlett, 2015, 716 CrossRef CAS.
- CCDC 1064109.
- For selected examples, see: (a) C. Liu, D. Liu, W. Zhang, L. Zhou and A. Lei, Org. Lett., 2013, 15, 6166 CrossRef CAS PubMed; (b) X. Wu, J. W. T. See, K. Xu, H. Hirao, J. Roger, J.-C. Hierso and J. S. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13573 CrossRef CAS PubMed; (c) T. Nishikata, Y. Noda, R. Fujimoto and T. Sakashita, J. Am. Chem. Soc., 2013, 135, 16372 CrossRef CAS PubMed.
- This pathway is also favoured on steric grounds when substrate bearing a methyl group at the 2-position of the aromatic ring was used (see ESI†).
- J. J. Devery III, J. J. Douglas, J. D. Nguyen, K. P. Cole, R. A. Flowers II and C. R. J. Stephenson, Chem. Sci., 2015, 6, 537 RSC.
- (a) E. Ferrer Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133, 10161 CrossRef PubMed; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details. CCDC 1064109. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc03951g |
| This journal is © The Royal Society of Chemistry 2015 |