
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe crystalline sponge method: MOF terminal ligand effects†
Timothy R.
Ramadhar
a,
Shao-Liang
Zheng
b,
Yu-Sheng
Chen
c and
Jon
Clardy
*a
aDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 240 Longwood Avenue, Boston, Massachusetts 02115, USA. E-mail: jon_clardy@hms.harvard.edu; Fax: +1-617-432-3790
bDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, Massachusetts 02138, USA
cChemMatCARS, Center for Advanced Radiation Sources, The University of Chicago c/o Advanced Photon Source, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, Illinois 60439, USA
First published on 2nd June 2015
Abstract
Bromide and chloride analogs of the commonly used zinc iodide-based metal organic framework for the crystalline sponge method were synthesized and evaluated. Inclusion of (1R)-(−)-menthyl acetate into these MOFs was analysed using third-generation synchrotron radiation, and the effects and potential benefits of varying the MOF terminal ligand are discussed.
The applicability of single crystal X-ray diffraction (SC-XRD) for structural analysis is limited by its requirement of a single crystal of the material to be analysed. Fujita and co-workers have recently described a method that could bypass this requirement by using a crystal of a metal organic framework (MOF) to orient host molecules within its pores. Their approach is called the crystalline sponge method,1 and the most widely used MOF sponge is {[(ZnI2)3(tris(4-pyridyl)-1,3,5-triazene)2]·x(solvent)}n (1a) (Fig. 1).2 In addition to providing a crystalline host for difficult-to-crystallize guests, this sponge’s heavy atoms allow for the absolute stereochemical determination of chiral guests with only light atoms through anomalous dispersion using Mo Kα radiation. The original report was met with enthusiasm3 and subsequent studies illustrated its utility,4 but the approach’s crystallographic quality, as highlighted by the incorrect stereochemical determination of miyakosyne A,5 has also been criticized.6 We previously reported an improved MOF synthetic procedure and the establishment of crystallographic guidelines that were derived from probing the systems with high-flux synchrotron radiation.7
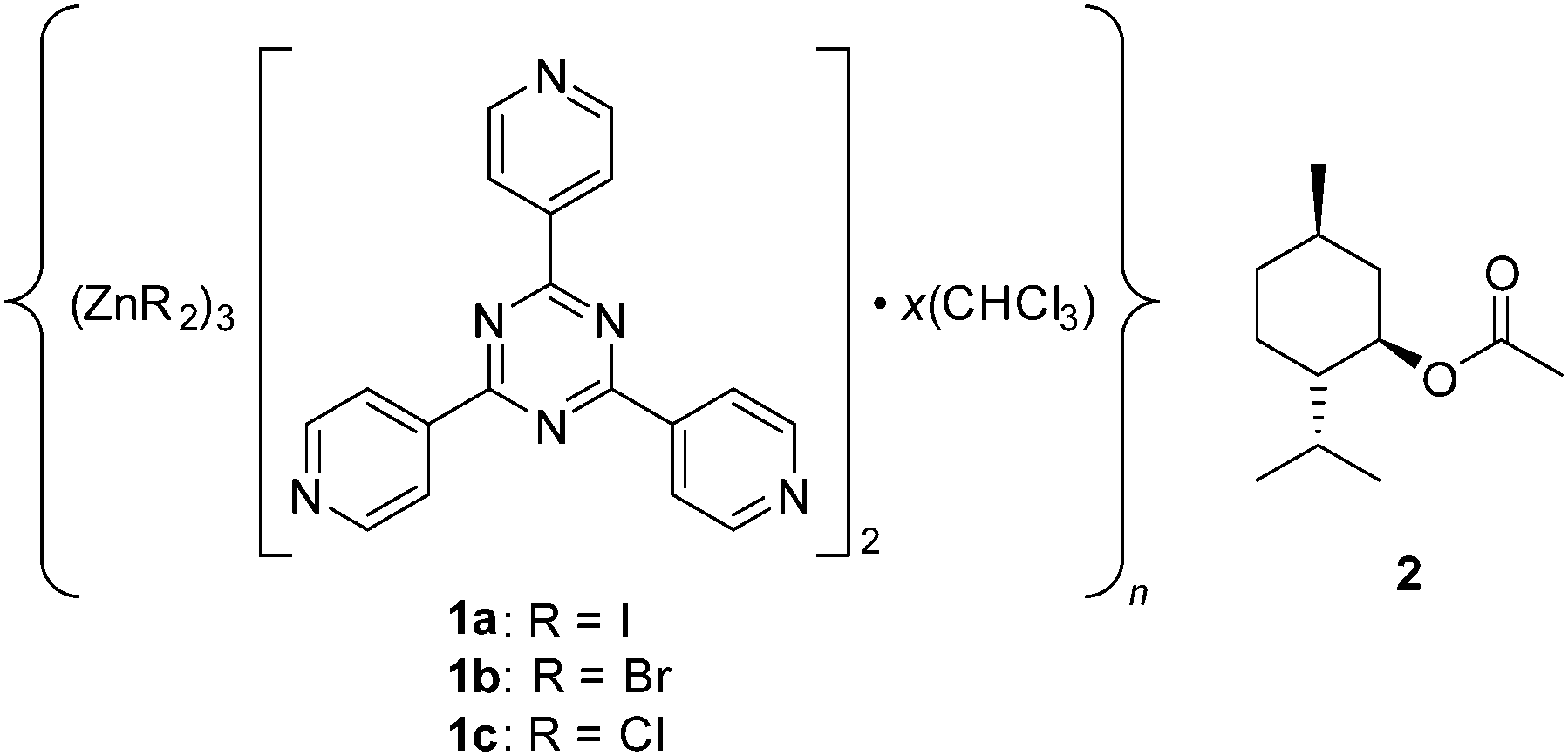 | ||
| Fig. 1 Crystalline sponges with various terminal halide ligands and the (1R)-(−)-menthyl acetate guest employed in the current study. | ||
Thus far, all prior crystalline sponge reports have typically used a zinc-based MOF with an iodide terminal ligand (1a).1,4,7 The crystallographic data to be analysed contain contributions from both the host and the guest, and the host makes the greatest contribution. We were interested in knowing whether changing the terminal ligand from iodide to bromide (1b)8 or chloride (1c)8,9 would simplify locating and modelling guest molecules since the lower electron count for bromide and chloride would increase the relative contribution of the guest, and other benefits would generally be imparted such as the reduction in X-ray absorption and lower diffuse scattering contribution to the background. Herein we communicate the synthesis, effects, and potential benefits of varying the crystalline sponge terminal ligand.
MOF crystals 1b and 1c were synthesized through a derivative procedure,7 where ZnBr2 and ZnCl2, respectively, were used in place of ZnI2. The crystals were grown in a bilayer of CHCl3 and MeOH for 7 days in order to maximize the crystal yield. This procedure led to the synthesis of high-quality MOF crystals and yielded a higher proportion of prismatic crystals versus those grown using ZnI2 where more thin sheets and twinned microcrystals were present.7 Crystals of 1b and 1c with CHCl3 solvent (“blank crystals”) exhibited weaker diffraction compared to the analogous crystals of 1a; thus, use of high-flux synchrotron radiation is beneficial for full data collection. Unit cell measurements for the 1b and 1c blank crystals were obtained on an in-house diffractometer, and in a similar manner to the 1a blank crystals,71b and 1c exhibited a centrosymmetric monoclinic C2/c space group and the unit cell dimensions were also similar.
We then performed the inclusion experiment with chiral target 1R-(−)-menthyl acetate (2), which was also used in our previous study with 1a to afford host–guest complex 1a2 (Fig. 2a).7 The soaking experiment for the 1b and 1c blank crystals with 2 was performed in the same manner as with the 1a blank crystals, where a neat guest was added to submerge the crystals for 2 d at ambient temperature. The resulting host–guest complexes 1b2 and 1c2 were then subjected to SC-XRD using high-flux synchrotron radiation with an irradiation wavelength of ∼0.41 Å (as an aside, X-ray fluorescence was also observed). In our original study in which 2 was introduced into 1a, the unit cell c-axis of the sponge approximately doubled to 66.990(6) Å and the space group symmetry decreased from C2/c to non-centrosymmetric monoclinic P21 affording 601 non-H atoms in the asymmetric unit including disorder (1a2).7 However, inclusion of 2 within 1b and 1c did not lead to a similar unit cell expansion. Furthermore, the space group symmetry decreased less as non-centrosymmetric monoclinic C2 for 1b2 and 1c2 was afforded versus P21 for 1a2. It should be noted that the combined figure of merit (CFOM) based on the statistical distribution of the normalised structure factors (〈|E2 − 1|〉) for 1b2 slightly favoured the space group choice of C2/c versus C2; thus, it was necessary to force data processing in the latter space and respective point group.
 | ||
| Fig. 2 Asymmetric unit for host–guest complexes (a) 1a2 containing 601 non-H atoms (ref. 7, CCDC 1007930), (b) 1b2 containing approximately 140 non-H atoms (CCDC 1063685), and (c) 1c2 containing approximately 142 non-H atoms (CCDC 1063686). | ||
The combined result of not expanding the unit cell and a higher space group symmetry led to asymmetric units containing approximately 140 and 142 non-H atoms for 1b2 and 1c2, respectively, versus 601 non-H atoms for 1a2,7 with all cases involving disorder (Fig. 2). The quantity of combined geometric and anisotropic displacement parameter restraints invoked for 1b2 and 1c2 is 95 and 80, respectively, versus 1137 for 1a2. The absolute configuration parameters based on anomalous dispersion for 1b2 and 1c2 unambiguously confirm the absolute stereochemistry of 2 (1b2: Flack x = 0.054(19), Hooft y = 0.026(19); 1c2: Flack x = 0.05(2), Hooft y = 0.031(19)).10,11 Furthermore, solvent accessible void electron density was not treated in 1b2 or 1c2 using PLATON/SQUEEZE12 or the Olex2 solvent mask.13 While the use of programs such as PLATON/SQUEEZE to treat residual density in solvent accessible voids affords lower refinement statistics in these systems, the analogous statistics without treating the residual density pass validation tests (no level A/B checkCIF alerts were afforded in this regard). Furthermore, previously reported guidelines for the crystalline sponge method state that a more realistic chemical representation of the system is afforded if residual density is not treated and that the use of these programs on these systems are best avoided if the R1 and wR2 refinement statistics are reasonable.7 Residual density from highly-disordered guest and solvent molecules is expected for the crystalline sponge systems by the nature of the method.
In both 1b2 and 1c2, two partially occupied guest molecules of 2 and residual chloroform solvent molecules (two in 1b2, one in 1c2) were observed in the asymmetric unit whereas 14 partially occupied targets 2 and one residual chloroform were observed in 1a2.7 Disorder of the terminal halide and zinc was observed and modelled on the host framework of 1b2 and 1c2. The R1 and wR2 refinement statistics for 1b2 (R1 = 7.10%, wR2 = 22.98%) and 1c2 (R1 = 8.47%, wR2 = 27.30%) are comparable to that for 1a2 (R1 = 6.19%, wR2 = 19.09%) albeit slightly larger. Since the host framework in 1b2 and 1c2 has proportionally lower contribution to the structure factors compared to 1a2, guest and residual solvent electron density is more readily observed, and modelling of the well-defined guests was straightforward with minimal soft restraints. However, residual electron density from severely disordered guest and solvent molecules, which is readily visible but cannot be reasonably modelled, as per previously reported guidelines,7 will inflate the R1 and wR2 refinement statistics, and leads to slightly larger absolute configuration parameters. Conversely, proportionally greater scattering contributions from the host in 1a2 originating from the higher electron count of the iodine atoms lead to highly disordered guest and residual solvent becoming more easily lost.
The differences in single-crystal-to-single-crystal transformation14 for 1a, 1b, and 1c upon inclusion of 2 is particularly intriguing. Unit cell expansion leading to 1a2 yielded a challenging scenario that required ample computational resources to perform least-squares refinement and weeks from initial solution to a structure ready for publication,7 which is highly undesirable from a practical standpoint. Moreover, long unit cell axes present a well-known crystallographic challenge that can ultimately affect data quality.15 Larger detector distances are required to resolve overlapping reflections; however, this leads to substantially weaker reflection intensities, especially in the high-angle shells. Use of longer irradiation wavelengths is an alternative strategy; however, this can make absorption correction difficult if strong absorbers such as iodide are present (and further exacerbated if the crystal habit is a plate or needle), and requires longer collection time to achieve sufficient completeness and redundancy due to a smaller sphere of reflection, spaced reflections, and detector size limitations requiring additional scans with multiple detector offsets. In the case of 1b2 and 1c2, the lack of both major unit cell expansion and reduction to P21 symmetry allowed a refinement time of a few hours instead of weeks. The exact origin of this effect in 1b2 and 1c2 is unclear. Since all datasets were collected using synchrotron radiation, determination of systematically absent reflections should be reliable. Visual comparison of the pores within all complexes after guest inclusion shows similar pore geometries albeit with some differences (Fig. 3), and void calculation using CalcVoid in Olex213 indicates that the structural components occupy a similar percentage within the unit cell (1a2: 36.52%, 1b2: 35.97%, 1c2: 35.35%). It is possible that inclusion of 2 within 1b and 1c induces a structural framework change that does not necessitate unit cell expansion and a drop to P21 symmetry.
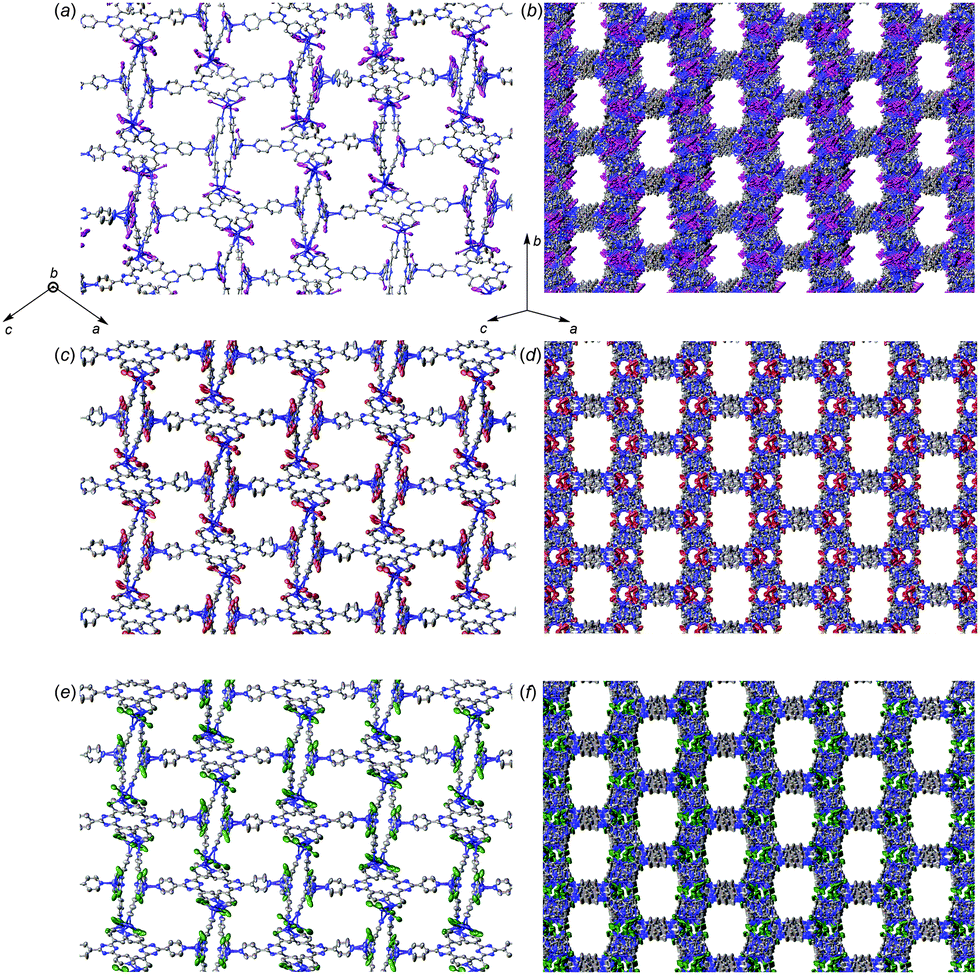 | ||
| Fig. 3 Views of the pores from different orientations within 1a2 (a), (b) (directly reproduced from ref. 7), 1b2 (c), (d), and 1c2 (e), (f). Hydrogen atoms and guest molecules are hidden for clarity. | ||
As the crystalline sponge method undergoes further refinements, a small library of generally but not universally useful sponges will emerge, and this report highlights the utility of varying the terminal ligand. The presented halide analogues can be easily prepared using the method described here, and their ability to affect both the single-crystal-to-single-crystal transformation upon introducing the guest and the concomitant data collection and analysis strategies have been discussed.
We are grateful for financial support through the US National Institutes of Health (U19-AI109673 to J.C. and F32-GM108415 to T.R.R.). ChemMatCARS Sector 15 is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1346572. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02-06CH11357.
Notes and references
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461 CrossRef CAS PubMed.
- K. Biradha and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3392 CrossRef CAS.
- (a) P. Stallforth and J. Clardy, Nature, 2013, 495, 456 CrossRef CAS PubMed; (b) E. Callaway, Nat. News, 2013, http://www.nature.com/news/taking-the-crystals-out-of-X-ray-crystallography-1.12699; (c) B. Halford, Chem. Eng. News, 2013, http://cen.acs.org/articles/91/i13/Crystal-Free-Crystallography.html.
- (a) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai and M. Fujita, Nat. Protoc., 2014, 9, 246 CrossRef CAS PubMed; (b) E. V. Vinogradova, P. Müller and S. L. Buchwald, Angew. Chem., Int. Ed., 2014, 53, 3125 CrossRef CAS PubMed; (c) A. G. O’Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868 CrossRef PubMed; (d) S. Yoshioka, Y. Inokuma, M. Hoshino, T. Sato and M. Fujita, Chem. Sci., 2015 10.1039/C5SC01681A; (e) V. Richards, Chem. World, 2015, http://www.rsc.org/chemistryworld/2015/05/crystal-free-X-ray-crystallography-axial-planar-chirality.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 501, 262 CrossRef CAS PubMed.
- (a) E. Callaway, Nat. News, 2013, http://www.nature.com/news/revolutionary-method-for-probing-molecular-structure-unravels-1.13798; (b) B. Halford, Chem. Eng. News, 2013, http://cen.acs.org/articles/91/i38/Crystal-Free-Crystallization-Loses-Lustre.html; (c) A. Extance, Chem. World, 2014, http://www.rsc.org/chemistryworld/2014/05/mechanism-study-seeks-clear-X-ray-crystalline-flask-cloud; (d) B. Halford, Chem. Eng. News, 2015, http://cen.acs.org/articles/93/i7/Crystalline-Sponges-Catching-Chemists.html.
- T. R. Ramadhar, S.-L. Zheng, Y.-S. Chen and J. Clardy, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 46 CAS.
- J. Martí-Rujas, N. Islam, D. Hashizume, F. Izumi, M. Fujita and M. Kawano, J. Am. Chem. Soc., 2011, 133, 5853 CrossRef PubMed.
- S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef.
- S. Parsons, H. D. Flack and T. Wagner, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 249 CAS.
- R. W. W. Hooft, L. H. Straver and A. L. Spek, J. Appl. Crystallogr., 2008, 41, 96 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- J.-P. Zhang, P.-Q. Liao, H.-L. Zhou, R.-B. Lin and X.-M. Chen, Chem. Soc. Rev., 2014, 43, 5789 RSC.
- Z. Dauter, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 1703 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and crystallographic procedures, and data tables. CCDC 1063685 and 1063686. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc03840e |
| This journal is © The Royal Society of Chemistry 2015 |