
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective electrochemical reduction of CO2 to CO with a cobalt chlorin complex adsorbed on multi-walled carbon nanotubes in water†
Shoko
Aoi
a,
Kentaro
Mase
a,
Kei
Ohkubo
ab and
Shunichi
Fukuzumi
*abc
aDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370
bDepartment of Bioinspired Science, Ewha Womans University, Seoul 120-750, Korea
cFaculty of Science and Technology, Meijo University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Nagoya, Aichi 468-8502, Japan
First published on 14th May 2015
Abstract
Electrocatalytic reduction of CO2 occurred efficiently using a glassy carbon electrode modified with a cobalt(II) chlorin complex adsorbed on multi-walled carbon nanotubes at an applied potential of −1.1 V vs. NHE to yield CO with a Faradaic efficiency of 89% with hydrogen production accounting for the remaining 11% at pH 4.6.
Electrocatalytic two-electron reduction of CO2 to CO has merited significant interest, because CO can be converted to liquid hydrocarbon fuels using H2 by Fischer–Tropsch processes.1–3 There have been extensive studies on the electrocatalytic reduction of CO2 with cobalt and nickel macrocycles.4–6 The selective electrocatalytic reduction of CO2 to CO has been achieved using nickel macrocycles in water without the formation of H2.7–9 Cobalt macrocycles can also act as good electrocatalysts for selective CO2 reduction to CO in organic solvents.10–13 In water, however, cobalt macrocycles have lacked the selectivity for CO,13–15 because cobalt complexes act as good catalysts for H2 evolution.16–21
We report herein the selective electrocatalytic reduction of CO2 to CO using a glassy carbon electrode modified with a cobalt(II) chlorin complex (CoII(Ch): a chemical structure shown in Scheme 1) adsorbed on carbon nanotubes in water.
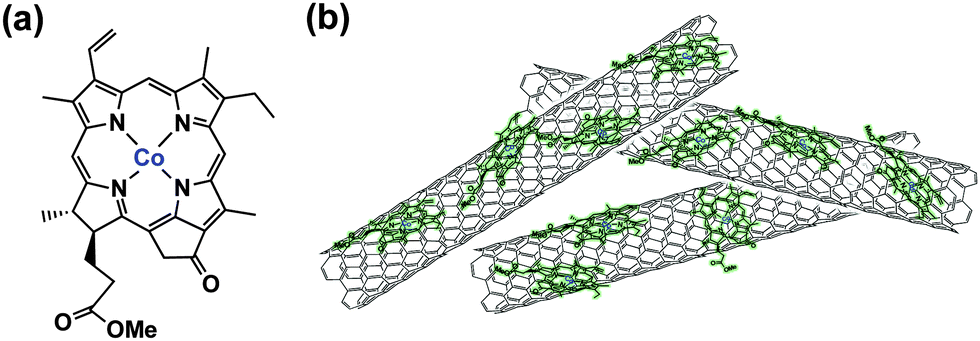 | ||
| Scheme 1 (a) Structure of CoII(Ch) and (b) schematic image of CoII(Ch) on MWCNTs. | ||
The CoII(Ch)-modified electrode was prepared by drop casting a sonicated acetonitrile (MeCN) solution containing CoII(Ch) (1.0 mM), multi-walled carbon nanotubes (MWCNTs: 1.3 mg) as a support material and 5% Nafion (12 μL) as a proton exchange membrane and stabilization agent of CoII(Ch) on MWCNTs to a glassy carbon electrode (experimental details are shown in the ESI†). Similarly the CoII(OEP)-modified electrode (OEP2− = octaethylporphyrin dianion) was prepared by sonication in an MeCN solution containing CoII(OEP) (1.0 mM), MWCNT and Nafion. The CoII(Ch)-modified electrode exhibited a catalytic current at an applied potential of <−1.0 V vs. NHE in a CO2-saturated aqueous solution at pH 4.6 as shown in Fig. 1 (red line). When CoII(Ch) was replaced by CoII(OEP) under otherwise the same experimental conditions, a decrease of the catalytic current from 60 μA (red line) to 30 μA (green line) at −1.1 V vs. NHE was observed as shown in Fig. 1.
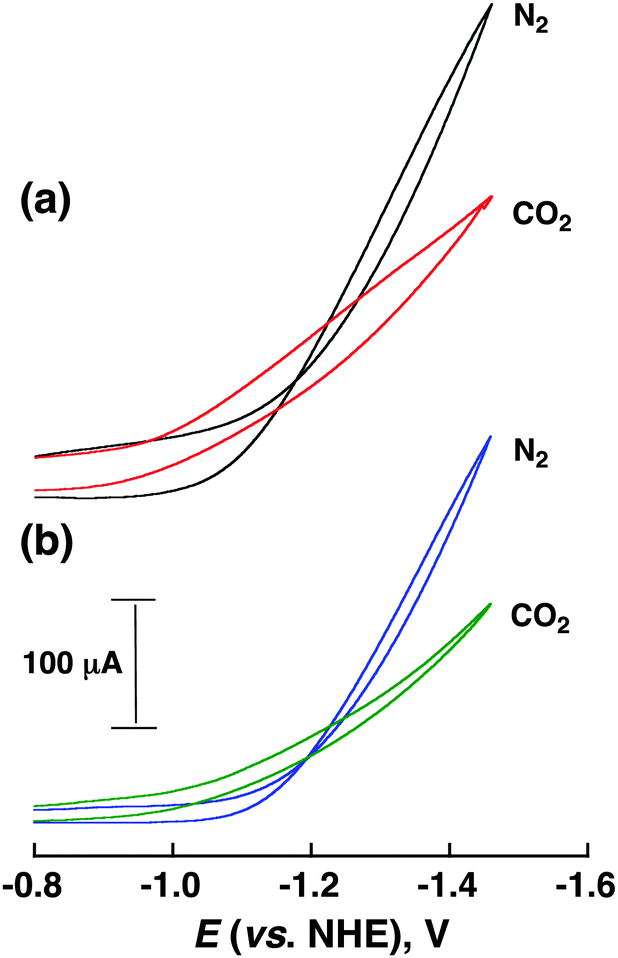 | ||
| Fig. 1 Cyclic voltammograms (CV) of (a) a CoII(Ch)-modified and (b) a CoII(OEP)-modified electrode in CO2- and N2-saturated aqueous solutions containing Na2SO4 (5.0 mM, pH = 4.6). Sweep rate: 10 mV s−1; working electrodes were modified with Co complex (0.01 μmol) and MWCNTs (13 μg) on a glassy carbon disk electrode. | ||
To assess the catalytic activity of CoII(Ch), controlled-potential electrolysis of a CO2-saturated aqueous solution with Na2SO4 (5.0 mM) as an electrolyte was performed and the formation of CO and H2 was quantitated by the gas chromatography analyses. No formation of the reduced products such as formaldehyde, methane, methanol and oxalate was observed under the present experimental conditions; however, a small amount of formic acid was detected by the formate dehydrogenase assay (Fig. S1 in the ESI†). We investigated various experimental conditions such as the pH of a CO2-saturated aqueous solution, an applied potential and various amounts of CoII(Ch) adsorbed on MWCNTs in a sonicated MeCN solution as summarized in Table 1. The time courses of formation of CO and H2 in electrolysis of a CO2-saturated aqueous solution with Na2SO4 at various pH values are shown in Fig. S2 in the ESI.† The maximum turnover number (TON) was 1100 with a TOF of 140 h−1 at pH 4.6. When the pH value is smaller than 4.6, proton reduction to evolve H2 occurred preferentially rather than CO2 reduction. The reason for a small TON at pH 6.8 is due to the slow proton-coupled electron-transfer reduction of CO2 to CO under high pH conditions. An applied potential and the concentration of CoII(Ch) for the CO formation were optimised to be −1.1 V vs. NHE and 1.0 mM, respectively (Fig. S3 and S4 in ESI†). Fig. 2 shows the time courses of formation of CO and H2 in electrolysis of a CO2-saturated aqueous solution with Na2SO4 under optimised conditions (i.e. pH 4.6, −1.1 V vs. NHE, [CoII(Ch)] = 1.0 mM), indicating that the CO yield is significantly higher than the H2 yield with the maximum turnover number (TON) of 1500 and a TOF of 100 h−1. The time courses of formation of CO and H2 in N2-saturated aqueous solution are shown in Fig. S5 in the ESI.† GC data are shown in Fig. S6 in the ESI.† The CVs and time courses of evolution of H2 under N2 and CO2 in the absence of CoII(Ch) or MWCNTs are shown in Fig. S7 and S8 in the ESI† as control experiments. No catalytic current for CO2 reduction and CO formation in the electrolysis was observed without MWCNTs or CoII(Ch). The current efficiency for CO production for the initial 2 h was determined to be as high as 89%, whereas that for H2 production was 11%.23 In the case of the CoII(OEP)-modified electrode, the selectivity for CO production decreased to 50% and the TON for CO production at 2 h of electrolysis was only 20 (Fig. S9 in the ESI†). Thus, the CoII(Ch)-modified electrode exhibits much higher electrocatalytic reactivity and selectivity for CO production than the CoII(OEP)-modified electrode.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: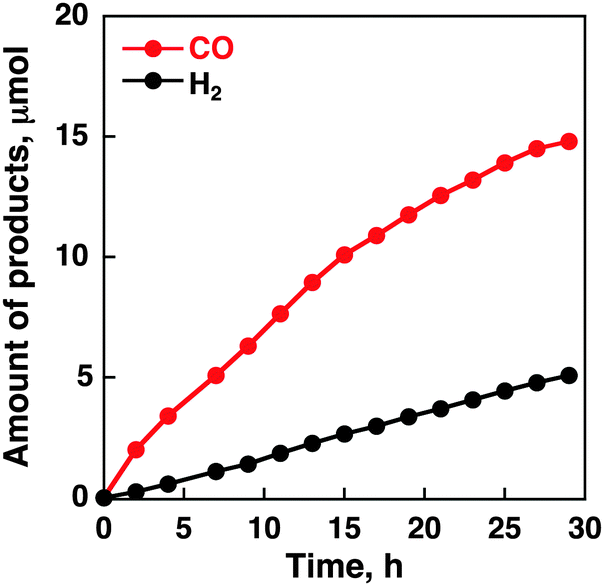 | ||
| Fig. 2 Time courses of evolution of CO and H2 in the electrocatalytic reduction of CO2 on the glassy carbon electrode modified with CoII(Ch) (0.01 μmol) adsorbed on MWCNTs (13 μg) in a CO2-saturated aqueous solution containing Na2SO4 (5.0 mM) at an applied potential of −1.1 V vs. NHE. CO2 was bubbled every 2 h. | ||
The EPR spectra of CoII(Ch) in a solution and CoII(Ch) on MWCNTs are shown in Fig. 3 to observe the π–π interaction between CoII(Ch) and MWCNTs. An EPR spectrum of a frozen MeCN solution containing CoII(Ch) at 4.2 K exhibited well-resolved signals at g = 2.293 (Fig. 3a), which is a typical low-spin five-coordinated cobalt(II) complex.24,25 On the other hand, an EPR spectrum of CoII(Ch) on MWCNTs showed a new signal at g = 4.203 in addition to the signal at g = 2.293 to the low-spin Co(II). The g = 4.203 signal is a triplet marker of two molecules of cobalt(II) complexes (S = 1/2) located close to each other. This indicates that the selective reduction of CO2 to CO results from involvement of two cobalt(I) complexes for two-electron transfer reduction of CO2.26
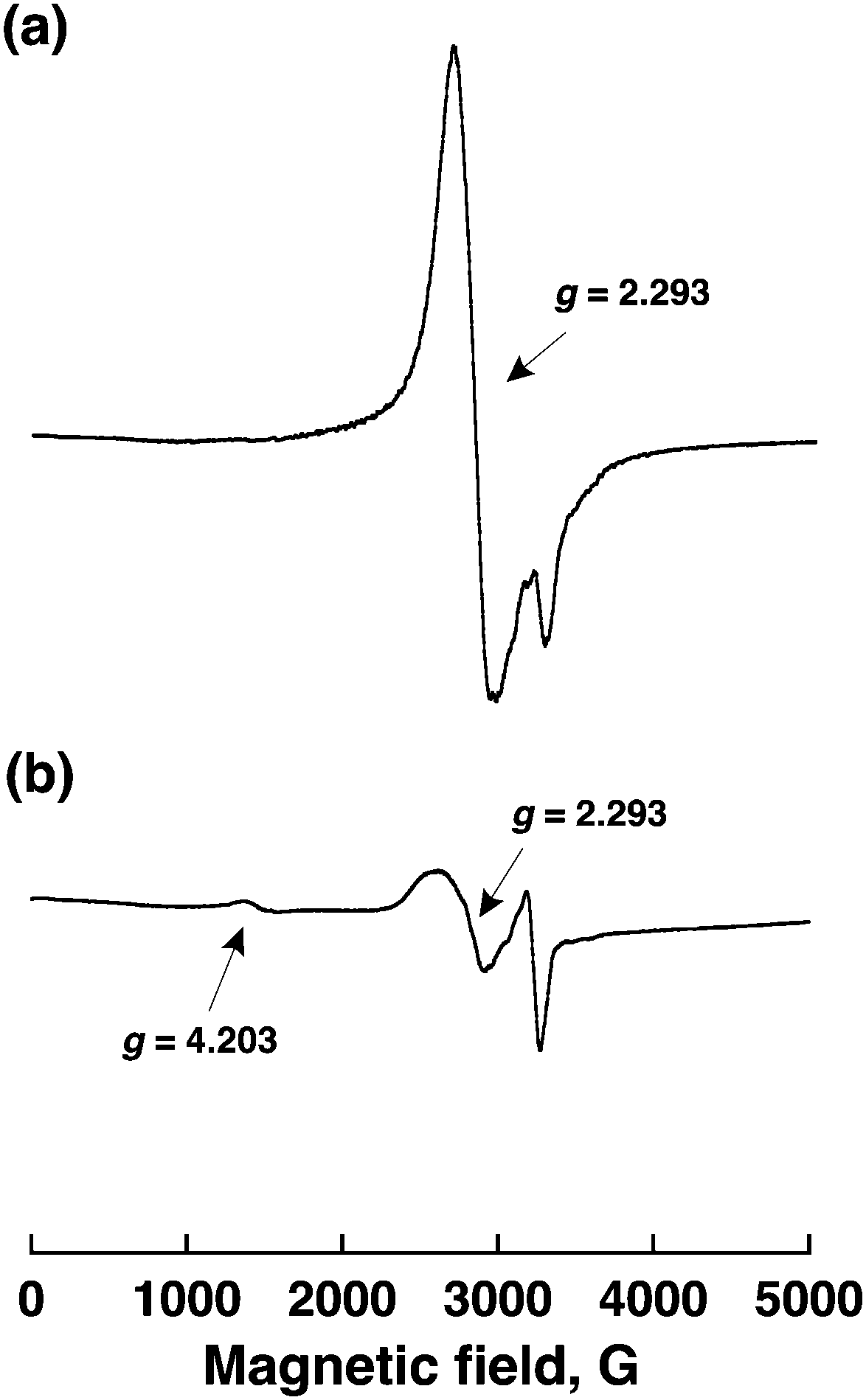 | ||
| Fig. 3 EPR spectra of (a) CoII(Ch) (1.0 mM) in deaerated MeCN measured at 4.2 K and (b) CoII(Ch) (1.0 mM) adsorbed on MWCNTs (0.30 mg) and Nafion (3.0 μL) in deaerated MeCN (250 μL) measured at 4.2 K. | ||
We also investigated the XPS measurements to confirm the state of the cobalt complex on MWCNTs after electrolysis. The XPS signal of the binding energy at 781 eV due to the Co(2p3/2) was shifted to 779 eV after electrolysis (Fig. S10 in the ESI†). The lower energy shift may be attributed to the reduction of Co(II) to form the low valent cobalt species.27 Thus, the deactivation of CoII(Ch) on MWCNTs may be attributed to the formation of unreactive monomer cobalt(I) species after the electrolysis.
When MWCNTs were replaced by reduced graphene oxide (rGO), which is planar, as a support material of CoII(Ch) (preparation procedures are shown in the Experimental section in the ESI†), the CO and H2 yields became much smaller (TON = 350 for CO and 250 for H2) (Fig. 4). Thus, the three dimensional assembly of MWCNTs with CoII(Ch) (Scheme 1b) on the electrode surface may play an important role for the selective electrocatalytic reduction of CO2 to CO. The π–π interaction between MWCNTs and CoII(Ch) also provides a suitable hydrophobic environment for binding of CO2 instead of proton, because the binding of CO2 to the Co(I) complex is required for the formation of CO.22 Because another Co(I) complex is also required for the two-electron reduction of CO2,22a two CoI(Ch) molecules located close to each other on MWCNTs facilitate CO2 reduction to CO (Scheme 1). Such situations may not be attained by a large two-dimensional π-system such as rGO, which afforded inefficient electrocatalytic reactivity for CO2 reduction with CoII(Ch).
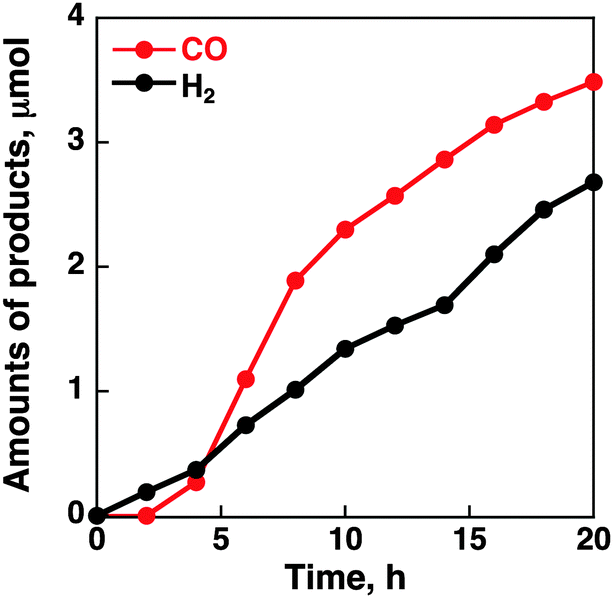 | ||
| Fig. 4 Time courses of formation of CO and H2 in the electrocatalytic reduction of CO2 on the glassy carbon electrode modified with CoII(Ch) (0.01 μmol) adsorbed on rGO (13 μg) in a CO2-saturated aqueous solution containing Na2SO4 (5.0 mM) at an applied potential of −1.1 V vs. NHE. | ||
In conclusion, a cobalt(II) chlorin complex adsorbed on MWCNTs acts as an efficient catalyst for selective electrocatalytic reduction of CO2 to CO in H2O (pH = 4.6) at an applied potential of −1.1 V vs. NHE with a high faraday efficiency of 89%. The present study provides a unique strategy for the selective electrocatalytic reduction of CO2 to CO over proton reduction to H2.
This work was supported by Grants-in-Aid (No. 26620154 and 26288037 to K.O.) and JSPS fellowship (No. 25727 to K.M.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT); ALCA and SENTAN projects from JST, Japan (to S.F.).
Notes and references
- P. Kang, Z. Chen, M. Brookhart and T. J. Meyer, Top. Catal., 2015, 58, 30 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed.
- (a) I. Wender, Fuel Process. Technol., 1996, 48, 189 CrossRef CAS; (b) M. Gupta, M. L. Smith, J. J. Spivey and J. James, ACS Catal., 2011, 1, 641 CAS; (c) Z.-J. Wang, Z. Yan, C.-J. Liu and D. W. Goodman, ChemCatChem, 2011, 3, 551 CrossRef CAS PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621 CrossRef CAS PubMed.
- B. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361 CrossRef CAS.
- (a) C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423 RSC; (b) J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036 RSC.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631 RSC.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461 CrossRef CAS PubMed.
- J. Schneider, H. Jia, K. Kobiro, D. E. Cabelli, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2012, 5, 9502 CAS.
- D. C. Lacy, C. C. L. McCrory and J. C. Peters, Inorg. Chem., 2014, 53, 4980 CrossRef CAS PubMed.
- D. W. Shaffer, S. I. Johnson, A. L. Rheingold, J. W. Ziller, W. A. Goddard, III, R. J. Nielsen and J. Y. Yang, Inorg. Chem., 2014, 53, 13031 CrossRef CAS PubMed.
- D. Quezada, J. Honores, M. García, F. Armijo and M. Isaacs, New J. Chem., 2014, 38, 3606 RSC.
- N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635 RSC.
- C. M. Lieber and N. S. Lewis, J. Am. Chem. Soc., 1984, 106, 5033 CrossRef CAS.
- T. V. Magdesieva, T. Yamamoto, D. A. Tryk and A. Fujishima, J. Electrochem. Soc., 2002, 149, D89 CrossRef CAS PubMed.
- (a) J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995 CrossRef CAS PubMed; (b) B. D. Stubbert, J. C. Peters and H. B. Gray, J. Am. Chem. Soc., 2011, 133, 18070 CrossRef CAS PubMed.
- E. Anxolabéhere-Mallart, C. Costentin, M. Fournier, S. Nowak, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 6104 CrossRef PubMed.
- C. C. L. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164 CrossRef CAS PubMed.
- P. V. Bernhardt and L. A. Jones, Inorg. Chem., 1999, 38, 5086 CrossRef CAS PubMed.
- L. Chen, M. Wang, K. Han, P. Zhang, F. Gloaguen and L. Sun, Energy Environ. Sci., 2014, 7, 329 CAS.
- S. Mandal, S. Shikano, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 15294 CrossRef CAS PubMed.
- (a) E. Fujita, D. J. Szalda, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1988, 110, 4870 CrossRef CAS; (b) E. Fujita, L. R. Furenlid and M. W. Renner, J. Am. Chem. Soc., 1997, 119, 4549 CrossRef CAS; (c) E. Fujita and R. van Eldik, Inorg. Chem., 1998, 37, 360 CrossRef CAS.
- The current efficiency in this work is higher than the reported values using cobalt terpyridine, porphyrin and phthalocyanine complexes.14,15.
- (a) T. Honda, T. Kojima and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 4196 CrossRef CAS PubMed; (b) K. Mase, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 2800 CrossRef CAS PubMed.
- A signal in the high magnetic field at g = 1.964 in Fig. 3 is assigned to the Co(III) superoxo complex [CoIII(Ch)–O2˙−], which is generated by the electron-transfer oxidation of CoII(Ch) with a small amount of O2 of residual at low temperature. See: (a) D. Sazou, C. Araullo-McAdams, B. C. Han, M. M. Franzen and K. M. Kadish, J. Am. Chem. Soc., 1990, 112, 7879 CrossRef CAS; (b) J. P. Collman, K. E. Berg, C. J. Sunderland, A. Aukauloo, M. A. Vance and E. I. Solomon, Inorg. Chem., 2002, 41, 6583 CrossRef CAS PubMed.
- We have examined EPR measurements in the presence of a lower concentration of CoII(Ch) on MWCNTs. The EPR signal at g = 4.2 was significantly smaller than the case of a high concentration of CoII(Ch).
- J. M. Gottfried, K. Flwchtner, A. Kretschmann, T. Lukasczyk and H.-P. Steinrück, J. Am. Chem. Soc., 2006, 128, 5644 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-Vis absorption spectra for the detection of formic acid (Fig. S1) and time courses of evolution of CO and H2 under various conditions (Fig. S2–S9). XPS of the working electrode (Fig. S10). See DOI: 10.1039/c5cc03340c |
| This journal is © The Royal Society of Chemistry 2015 |