
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gating the photochromism of an azobenzene by strong host–guest interactions in a divalent pseudo[2]rotaxane†
Mirko
Lohse
a,
Karol
Nowosinski
b,
Nora L.
Traulsen
b,
Andreas J.
Achazi
b,
Larissa K. S.
von Krbek
b,
Beate
Paulus
*b,
Christoph A.
Schalley
*b and
Stefan
Hecht
*a
aDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: sh@chemie.hu-berlin.de; Fax: +49 (0)30 2093-6940; Tel: +49 (0)30 2093-7308
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustraße 3, 14195 Berlin, Germany. E-mail: b.paulus@fu-berlin.de; christoph@schalley-lab.de; Fax: +49 (0)30 838-55366; Tel: +49 (0)30 838-52639
First published on 1st May 2015
Abstract
The ability of an E-configured azobenzene guest to undergo photoisomerisation is controlled by the presence of a complementary host. Addition of base/acid allowed for a weakening/strengthening of the interactions in the divalent pseudo[2]rotaxane complex and hence could switch on/off photochromic activity.
Photochromism, the reversible switching of a molecule between two states by light, provides an excellent tool for the development of smart materials by gaining remote control over their properties via an external stimulus.1 Being able to turn on and off the ability of a photoswitch to isomerise, so-called “gated photochromism”, is desirable to implement responsiveness to multiple stimuli2 and has primarily been achieved by acid–base reactions or coordination of (transition) metal ions.3 In addition, specific host–guest interactions involving one isomer of the photoswitch selectively could in principle also be employed to gate the switching process and in turn provide photocontrol over the association process. The underlying binding event should be readily modulated by large structural changes in either the host or guest. Therefore, it seems reasonable to utilise a photoswitch, which undergoes a light-induced E–Z double bond isomerisation. In this context, azobenzene can be considered as the “drosophila” in the field. Its significant structural changes upon E–Z photoisomerisation have been used to modulate microscopic4 and even macroscopic5 property changes, which have been exploited in various areas from chemistry6 over material science1,5,7 to biology8 and potentially pharmacology.9 Whereas authors typically care about the influence of azobenzene photoisomerisation on the resulting supramolecular structure,10 much less attention has been paid to cases where the non-covalent interactions influence the azobenzene's photoswitchability.11
Here, we describe the first pseudo[2]rotaxane assembly involving a divalent crown ether host and a divalent photochromic azobenzene guest. By making or breaking of the host–guest complex we can either lock or unlock the E-isomer and thereby gate its photoisomerisation ability.
The key components of our host–guest system are photochromic axles 1 and 2, carrying two terminal secondary ammonium-binding sites, which are based on azobenzene and stilbene, respectively (Fig. 1). The latter has been prepared in particular to access Z-2 as a thermally stable, structural analogue of Z-1.12 Based on the well-known and tuneable interaction between a secondary ammonium ion and a dibenzo-24-crown-8,13 anthracene-spacered divalent crown ether 314 was chosen as the complementary divalent host. In addition, monovalent ammonium guest 5 and monovalent crown ether 4 were prepared for comparison purposes.
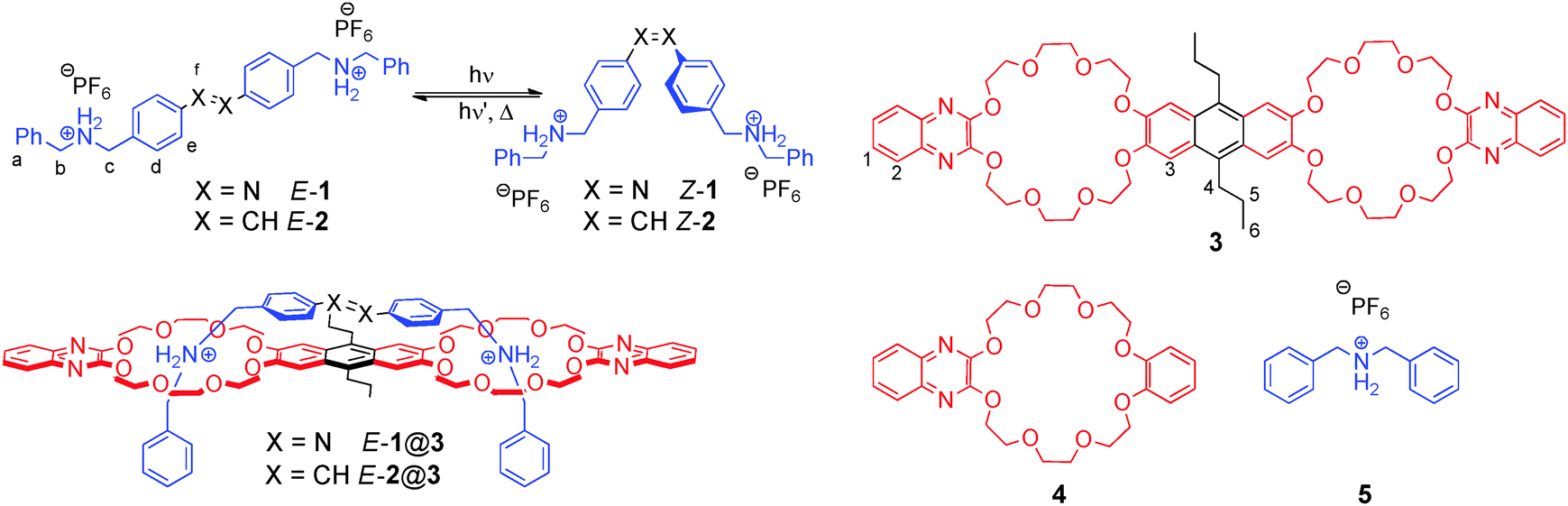 | ||
| Fig. 1 Isomerization of divalent azobenzene and stilbene axles 1 and 2, divalent host 3, monovalent reference compounds 4 and 5, as well as divalent pseudo[2]rotaxanes E-1@3 and E-2@3. | ||
Synthesis of the azobenzene axle 1 involves a straightforward linear sequence of reductive amination of 4-nitrobenzaldehyde with benzyl amine and BOC-protection, followed by reduction to the aniline derivative and oxidative dimerization. BOC-deprotection and protonation with NH4PF6 finally yields hexafluorophosphate salt E-1. Axles E-2 and Z-2 were derived from the corresponding (E)-15 and (Z)-stilbene16 dialdehydes via reductive amination and protonation. The synthesis and characterisation of host 3 have been described previously.14 More information about the synthesis and compound characterisation is provided in the ESI.†
To investigate the host–guest interactions between guest E-1 and host 3, 1H NMR spectroscopy experiments have been carried out. In an equimolar mixture, a new set of signals appears, which is related to complex E-1@3 (Fig. 2 top, see also ESI†, Fig. S1 and S2). Whereas significant chemical upfield shifts were observed for the aromatic protons Hd (Δδ ≈ 0.35 ppm) and He (Δδ ≈ 0.7 ppm) of the azobenzene core of axle E-1 due to their position atop the anthracene unit of host 3 indicating exclusive formation of doubly bound dimers. The benzylic protons Hb and Hc shifted downfield (Δδ ≈ 0.5 ppm) upon complexation. These large shifts can be explained by the location of the azobenzene protons above the anthracene core of host 3 on the one hand and the deshielding of the benzylic protons due to C–H⋯O hydrogen-bonding with the crown ether oxygen atoms as well as due to the aromatic ring current of the anthracene spacer, on the other. Note that the splitting of several peaks for crown ether methylene groups in the complex E-1@3 is caused by the threading of the axle through the host, resulting in new sets of diastereotopic proton signals. EXSY NMR studies show that in the chosen solvent mixture (CDCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CD3CN = 2:1) there is no detectable exchange between the bound and free axle E-1, indicating that dethreading is slow. In addition, ESI mass spectrometry supports the formation of the complex E-1@3 (see ESI†, Fig. S25).
:CD3CN = 2:1) there is no detectable exchange between the bound and free axle E-1, indicating that dethreading is slow. In addition, ESI mass spectrometry supports the formation of the complex E-1@3 (see ESI†, Fig. S25).
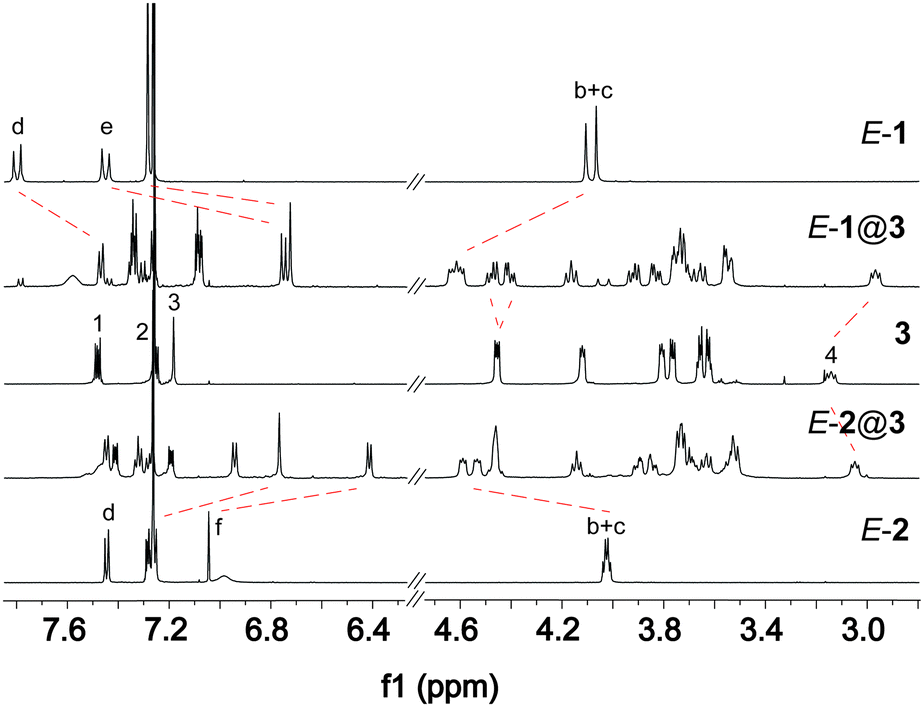 | ||
| Fig. 2
1H NMR experiment of the complexation of azobenzene axle E-1 (top) as well as E-2 (bottom) to host 3 (500 MHz, CDCl3:CD3CN 2:1, 1.4 mM). The dashed lines indicate the shifting of selected protons upon formation of the complex. | ||
Trying to elucidate the structure of the host–guest complex formed by the photoisomerised axle Z-1 is practically impossible due to its thermal Z → E back isomerisation (note that the inevitably forming E-1 binds to the host 3 much more strongly). Therefore, the isosteric stilbene axles E-2 and Z-2 have been studied. While 1H NMR titration reveals formation of a stable 1:1 complex E-2@3 (Fig. 2, bottom), the Z-configured axle Z-2 does not yield a well-defined complex upon addition of 3 (see ESI†, Fig. S27).
In order to investigate the association behaviour of the different axles with the host more thoroughly, a detailed thermodynamic analysis of the assembly was carried out involving isothermal titration calorimetry (ITC), which has proven very insightful for studying strongly bound pseudorotaxanes.17 In contrast to similar pseudo[2]rotaxanes based on crown ether–ammonium interactions,17 a very strong binding was observed for complex E-1@3, giving rise to an unusually high association constant of Ka = 200000 M−1 (Table 1). This strong interaction demonstrates the perfect fit of axle E-1 to host 3. Additional secondary binding events, such as acceptor–donor type π,π-stacking of the electron-deficient azobenzene and the electron-rich anthracene cores,18 together with the divalent complexation contribute to a large effective molarity (EM) value of 380 mM and a highly positive chelate cooperativity factor of Kmono × EM = 340 ≫ 1.19 Additional ITC experiments to deduce the association constants of the stilbene-based complexes E-2@3 and Z-2@3 were not successful due to insufficient solubility of the axle components. However, in an insightful competition experiment monitored by 1H NMR spectroscopy it could be shown that upon addition of E-1 to an equimolar solution of Z-2 and 3, E-1@3 was formed rapidly (see Fig. S30 in ESI†). This indicates that the association constant of Z-2@3 containing the Z-configured (stilbene) axle has to be much lower than the one of the complex composed of the E-configured (azobenzene) axle. The association constants of the monovalent model complexes 5@4, E-1@42 and 52@3 are all lower and in a similar range. Higher Ka values for the first binding step in case of E-1@42 and 52@3 compared to 5@4 are expected due to statistics. However, both show negative allosteric interaction19 with reduced Ka values for the second binding step and allosteric cooperativity factors α(E-1@42) = 0.8 and α(52@3) = 0.2, which has been found before for similar systems.17 We attribute the quite strong negative allosteric cooperativity of 52@3 to the polarization of the π-system of the crown ether's anthracene spacer by the first ammonium ion. This increases positive or diminishes negative partial charges at the second crown ether binding site and thus reduces the binding strength of the second monovalent axle.
:CH3CN = 2.2:1, 298 K)
| Complex | K a [M−1] | ΔG [kJ mol−1] | ΔH* [kJ mol−1] | TΔS*a [kJ mol−1] | |
|---|---|---|---|---|---|
| a ΔH and ΔS values have larger errors than those of ΔG and should be regarded as estimate rather than precise values. | |||||
| E-1@3 | 2 × 105 ± 2 × 104 | −30.3 ± 0.2 | −52.1 | −21.9 | |
| 5@4 | 1800 ± 200 | −18.6 ± 0.2 | −39.3 | −20.6 | |
| E-1@42 | K 1 | 3100 ± 300 | −19.9 ± 0.3 | −27.6 | −7.7 |
| K 2 | 870 ± 90 | −16.8 ± 0.2 | −35.0 | −18.1 | |
| 5 2@3 | K 1 | 3700 ± 400 | −20.4 ± 0.2 | −26.3 | −5.9 |
| K 2 | 170 ± 20 | −12.7 ± 0.3 | −32.9 | −20.2 | |
To gain further insight into the binding situation, in particular of the illusive complexes composed of the Z-configured axles, the Gibbs energies ΔG for the 1:1 association were calculated by DFT using an approach developed by Grimme.20 The calculations were performed with TPSS-D3(BJ)/def2-TZVP21 including solvent effects with the COSMO-RS solvent model.22 In these calculations it is assumed that one PF6− anion is close to the free guest and has to be moved away from the guest during the threading. We have recently described this approach in detail for similar systems and the computed results agree well with the experiment.23 For E-1@3 the calculated Gibbs energy of association ΔG (Table S2 in ESI†) is close to the experimental value, validating our method. According to these calculations it turns out that both divalent complexes Z-1@3 and Z-2@3 are thermodynamically unfavourable. Hence, they should not form in agreement with our experiments showing no evidence for the formation of discrete 1:1 complexes in the case of the Z-configured axles. Furthermore, the calculations predict a weaker yet favourable interaction energy for E-2@3, which is in line with a weaker π,π-stacking of the more electron-rich stilbene moiety but cannot be compared with experiment since no quantitative data are accessible. The strong stabilisation of the E-isomer upon binding to the host is furthermore evident from comparison of the Gibbs energies for the E-1 → Z-1 isomerisation in the free (ΔG = +28.8 kJ mol−1) and in the complexed (ΔG = +115.3 kJ mol−1) form (detailed description see ESI†).
After analysis of the binding of the axles in their two isomeric forms to the host, we were interested in the effect of this complexation on the photoisomerisation behaviour. In the absence of host 3, axle E-1 shows the typical spectroscopic signature of a regular azobenzene derivative with an intense π,π* band at 333 nm and a weak shoulder at 450 nm associated with the symmetry forbidden n,π* transition (Fig. 3, top). Upon irradiation at 334 nm, the π,π* band decreases while the n–π* band increases and a photostationary state (PSS) with 90% Z-1 is reached. The quantum yield of the E → Z photoisomerisation for axle 1 under these conditions is in the expected range ΦE→Z = (6.8 ± 0.1)%. The activation barrier for the thermal Z → E isomerisation of Z-1 amounts to ΔG‡ = (110 ± 7) kJ mol−1, giving rise to a thermal half-life of τ1/2 = (127 ± 1) h at room temperature (see ESI†, Fig. S33 and S34). When mixing axle E-1 and host 3, their absorption spectra largely overlap (for the absorption spectra of individual compounds see Fig. S38 in ESI†). To prevent potential photooxidation of the anthracene moiety,24 all photochemical experiments were carried out in degassed solvents. Upon irradiation of an equilibrated 1:1 mixture of the axle and host compounds at 334 nm, only minor spectral changes were observed, indicating only little photoisomerisation taking place in E-1@3. This gives evidence for the successful inhibition of azobenzene photochromism (Fig. 3, bottom).
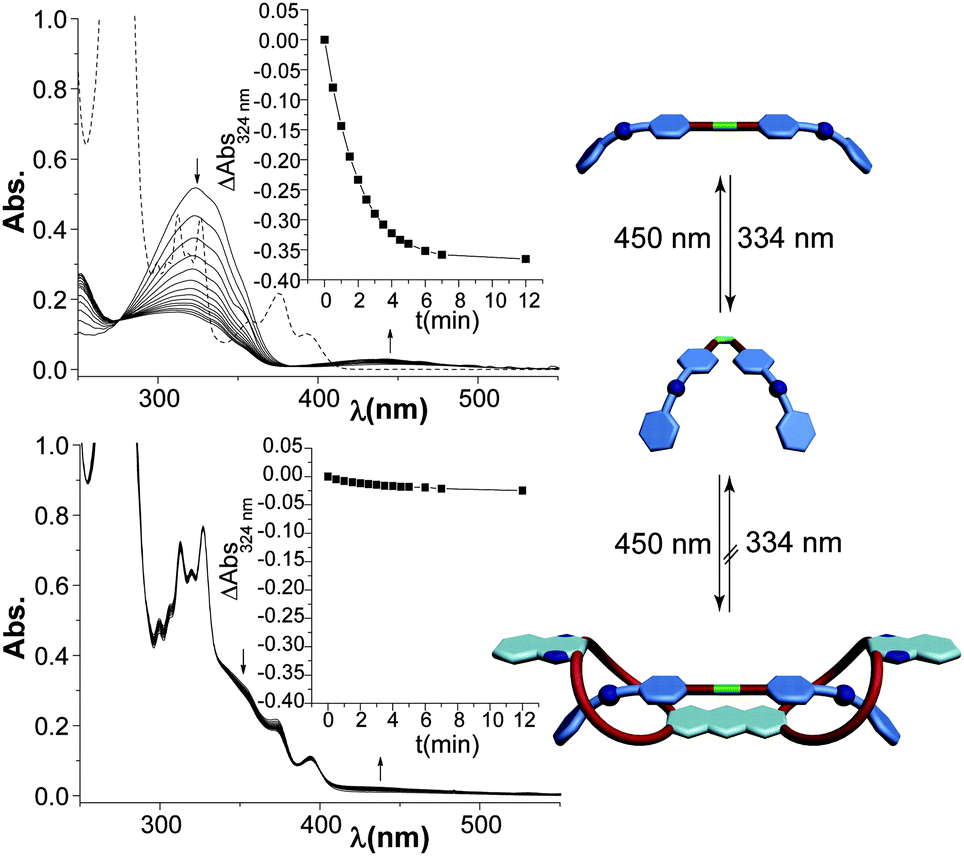 | ||
| Fig. 3
E → Z photoisomerisation of E-1 (40 µM in CHCl3:CH3CN = 2:1) upon irradiation with 334 nm in the absence (top) and presence (bottom) of an equimolar amount of host 3 (dashed line shows spectrum of pure 3). | ||
Binding to host 3 selectively inhibits the forward E → Z photoisomerisation of 1, but does not affect the reverse process since irradiation of a PSS mixture (containing 90% Z-1 and 10% E-1) in the presence of 3 at 436 nm showed clean Z → E photoisomerisation (see Fig. S37a in ESI†). However, the presence of host 3 does not accelerate the thermal Z → E isomerisation by selective stabilisation of E-1@3, but in contrast the aggregate formed with Z-1 decelerates the thermal back reaction (for comparison of thermal Z → E isomerisation in the free and complexed form see Fig. S35, ESI†). However, a clean E → Z photoisomerisation can be observed when irradiating E-1 in the presence of excess monovalent host 4, i.e. forming E-1@42 (see Fig. S36b in ESI†). This clearly shows that complexation of the ammonium ions is not responsible for the observed inhibition effect. Furthermore, it is possible to induce E → Z photoisomerisation of the dethreaded, unprotonated axle in the presence of 3 (see Fig. S36a in ESI†), showing that the overlapping spectra of both species are neither the reason for the inhibition. Fluorescence measurements (see ESI†, Fig. S39) of the E-1@3 complex show no indication of energy transfer at various excitation wavelengths but in contrast, complexation leads to fluorescence quenching of the host, presumably due to photoinduced electron transfer.25 In view of the strong binding in E-1@3 and considering the tuneability of the ammonium ion–crown ether interaction by solvent polarity, another switching experiment was carried out in a more polar solvent mixture, i.e. 14:1 instead of 1:2 CH3CN:CHCl3. In this case, the attractive interactions between axle E-1 and host 3 are reduced due to competition of their hydrogen bond acceptors and donors with the polar solvent. As a consequence, inhibition becomes much less effective and the photochromic behaviour is recovered (see ESI†, Fig. S37b). Note that the weaker binding under these conditions could not be quantified by ITC due to insufficient solubility in the acetonitrile-rich medium. All of the above experiments clearly indicate that formation of the E-1@3 complex is actually responsible for inhibition and suggest that in the tight, vice-like pseudo[2]rotaxane binding scenario E → Z photoisomerisation is shut down.
Comparing the E → Z photoisomerisation at these different conditions, an inhibition efficiency of the pseudo[2]rotaxane system can be estimated. Looking at the decay of the E-azobenzene absorption maximum at 350 nm, one can see that the decrease after 60 min for the complex E-1@3 is approximately 7 times slower than for the deprotonated and therefore non-complexed case, 1–2H+ + 3, as well as the monovalent host case, i.e. E-1@42 (Fig. 4, first 60 min). Subsequent in situ deprotonation of the binding sites in the E-1@3 complex quickly re-establishes photochromism and results in a fast E → Z photoisomerisation. Upon addition of trifluoroacetic acid to the fully converted mixture and irradiating with 436 nm light Z → E photoisomerisation takes place and the system is reset thereby showing the reversibility of the process. These experiments demonstrate gating of azobenzene photoisomerisation by addition of either base or acid to the pseudo[2]rotaxane host–guest system.
 | ||
| Fig. 4
E → Z photoisomerisation of E-1 in presence of 3 (1@3, solid black line), deprotonated E-1 in presence of 3 (1–2H+ + 3, dashed black line), and E-1 in presence of 4 (1@42, solid red line) monitoring azobenzene absorption maximum at 350 nm upon irradiation at 334 nm (40 µM in CHCl3:CH3CN = 2:1). After 60 min DBU (4 equiv.) was added to the solution of E-1@3 leading to deprotonation and recovery of photoreactivity. Subsequent Z → E photoisomerisation at 436 nm and addition of TFA (4.1 equiv.) reprotonates the axle and resets the system. Note that the system does not recover fully because of the photostationary state of deprotonated 1 and photochemical side reactions of 3. | ||
In the present study, we found a unique way in which strong binding of a divalent azobenzene guest to a complementary host was found to effectively inhibit E → Z photoisomerisation. This complexation-induced gating effect can be tuned by changing the strength of the non-covalent interactions, either by varying solvent polarity or more effectively by adding base or acid. On-going work is focusing on shifting the irradiation wavelengths further to the red to achieve a selective excitation of the azobenzene axle.12
The authors thank the Deutsche Forschungsgemeinschaft for generous financial support (SFB 765). Support by the High-Performance Computing facilities of the Freie Universität Berlin (ZEDAT) is acknowledged. L.v.K. is grateful to the Studienstiftung des Deutschen Volkes for a PhD fellowship.
Notes and references
- M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348 CrossRef CAS PubMed.
- In a different context this is related to the concept of molecular-scale logic gates, see: A. P. de Silva and N. D. McClenaghan, Chem. – Eur. J., 2004, 10, 574 CrossRef PubMed.
- J. Zhang, Q. Zhu and H. Tian, Adv. Mater., 2013, 25, 378 CrossRef CAS PubMed.
- (a) T. Hugel, N. B. Holland, A. Cattani, L. Moroder, M. Seitz and H. E. Gaub, Science, 2002, 296, 1103 CrossRef PubMed; (b) M. Alemani, M. V. Peters, S. Hecht, K.-H. Rieder, F. Moresco and L. Grill, J. Am. Chem. Soc., 2006, 128, 14446 CrossRef CAS PubMed; (c) C. Dri, M. V. Peters, J. Schwarz, S. Hecht and L. Grill, Nat. Nanotechnol., 2008, 3, 649 CrossRef CAS PubMed.
- H. Yu and T. Ikeda, Adv. Mater., 2011, 23, 2149 CrossRef CAS PubMed.
- (a) R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054 CrossRef CAS PubMed; (b) R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982 RSC.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139 CrossRef CAS PubMed.
- (a) A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422 RSC; (b) T. Fehrentz, M. Schönberger and D. Trauner, Angew. Chem., Int. Ed., 2011, 50, 12156 CrossRef CAS PubMed; (c) T. S. Zatsepin, L. A. Abrosimova, M. V. Monakhova, H. Le Thi, A. Pingoud, E. A. Kubareva and T. S. Oretskaya, Russ. Chem. Rev., 2013, 82, 942 CrossRef CAS PubMed.
- (a) W. Szymanski, J. M. Beierle, H. A. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114 CrossRef CAS PubMed; (b) W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178 CrossRef CAS PubMed.
- (a) X. Chen, L. Hong, X. You, Y. Wang, G. Zou, W. Su and Q. Zhang, Chem. Commun., 2009, 1356 RSC; (b) G. Wenz, B.-H. Han and A. Müller, Chem. Rev., 2006, 106, 782 CrossRef CAS PubMed; (c) D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015 DOI:10.1021/cr5006342; (d) S. Yagai, T. Karatsu and A. Kitamura, Chem. – Eur. J., 2005, 11, 4054 CrossRef CAS PubMed; (e) S. Hecht, Small, 2005, 1, 26 CrossRef CAS PubMed; (f) C. Stoffelen, J. Voskuhl, P. Jonkheijm and J. Huskens, Angew. Chem., Int. Ed., 2014, 53, 3400 CrossRef CAS PubMed.
- (a) M. S. Vollmer, T. D. Clark, C. Steinem and R. H. Ghadiri, Angew. Chem., Int. Ed., 1999, 38, 1598 CrossRef CAS; (b) M. Yamamura, Y. Okazaki and T. Nabeshima, Chem. Commun., 2012, 48, 5724 RSC; (c) H.-S. Tang, N. Zhu and V. W.-W. Yam, Organometallics, 2006, 26, 22 CrossRef.
- The thermal half-life of regular (Z)-azobenzenes is only on the time scale of minutes to few hours. For a notable recent exception, see: (a) D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597 CrossRef PubMed; (b) C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bléger, Chem. – Eur. J., 2014, 20, 16492 CrossRef CAS PubMed.
- (a) J. M. Timko, S. S. Moore, D. M. Walba, P. C. Hiberty and D. J. Cram, J. Am. Chem. Soc., 1977, 99, 4207 CrossRef CAS; (b) P. R. Ashton, P. J. Campbell, P. T. Glink, D. Philp, N. Spencer, J. F. Stoddart, E. J. T. Chrystal, S. Menzer, D. J. Williams and P. A. Tasker, Angew. Chem., Int. Ed., 1995, 34, 1865 CrossRef CAS PubMed; (c) W. Jiang, P. C. Mohr, A. Schäfer and C. A. Schalley, J. Am. Chem. Soc., 2010, 132, 2309 CrossRef CAS PubMed.
- W. Jiang and C. A. Schalley, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10425 CrossRef CAS PubMed.
- M. Linseis, S. Záli
![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) , M. Zabel and R. F. Winter, J. Am. Chem. Soc., 2012, 134, 16671 CrossRef CAS PubMed.
, M. Zabel and R. F. Winter, J. Am. Chem. Soc., 2012, 134, 16671 CrossRef CAS PubMed. - T. Bosanac and C. S. Wilcox, Org. Lett., 2004, 6, 2321 CrossRef CAS PubMed.
- (a) W. Jiang, K. Nowosinski, N. L. Löw, E. V. Dzyuba, F. Klautzsch, A. Schäfer, J. Huuskonen, K. Rissanen and C. A. Schalley, J. Am. Chem. Soc., 2011, 134, 1860 CrossRef PubMed; (b) L. Kaufmann, N. L. Traulsen, A. Springer, H. V. Schröder, T. Mäkelä, K. Rissanen and C. A. Schalley, Org. Chem. Front., 2014, 1, 521 RSC; (c) L. Kaufmann, E. V. Dzyuba, F. Malberg, N. L. Löw, M. Groschke, B. Brusilowskij, J. Huuskonen, K. Rissanen, B. Kirchner and C. A. Schalley, Org. Biomol. Chem., 2012, 10, 5954 RSC.
- C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2001, 651 RSC.
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488 CrossRef CAS PubMed.
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Eckert and A. Klamt, COSMOtherm, Version C3.0, Release 13.01, COSMOlogic GmbH & Co. KG Leverkusen, Germany, 2013 Search PubMed.
- A. J. Achazi, L. K. S. von Krbek, C. A. Schalley and B. Paulus, J. Comput. Chem., 2015 DOI:10.1002/jcc.23914.
- (a) E. J. Bowen and D. W. Tanner, Trans. Faraday Soc., 1955, 51, 475 RSC; (b) W. Jiang, M. Han, H. Y. Zhang, Z. J. Zhang and Y. Liu, Chem. – Eur. J., 2009, 15, 9938 CrossRef CAS PubMed.
- M. V. Peters, R. Goddard and S. Hecht, J. Org. Chem., 2006, 71, 7846 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data, ITC and photochemistry experiments. See DOI: 10.1039/c5cc02811f |
| This journal is © The Royal Society of Chemistry 2015 |