
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn aquo ppm level detection of acrylamide through S-to-N acyl transfer mediated activation of pro-sensors†
Amarendar
Reddy M
and
Aasheesh
Srivastava
*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhauri, Indore By-pass Road, Bhopal, 462 066, India. E-mail: asri@iiserb.ac.in
First published on 29th May 2015
Abstract
Water-soluble pro-sensors for acryl compounds including acrylamide (AM) were developed. Spontaneous conversion to active thiolate (Ph-S and Nap-S) occurs through pH-increase. While Ph-S forms flat nano-tapes with AM, Nap-S acted as a turn-on fluorescence detector, sensing AM up to the ppm level.
Different acrylic monomers such as acrylamide (AM) and its precursor acrylonitrile (AN), as well as acrylates such as methyl acrylate (MA), ethyl acrylate (EA) and methyl methacrylate (MM) are produced industrially in tonnage quantities. These monomers are employed as raw materials in plastic and textile industries.1,2AM is also employed as a flocculent in sewage treatment and as a soil conditioning agent. AM is also reported to be present in industrially prepared food items that have been heated above 120 °C,3 and its presence in biological fluids is carefully monitored due to potential negative health effects. The deleterious health effects of AM vary with the extent of exposure – while short term contact may result in severe eye and skin irritation, long term exposure may have carcinogenic effects.4–6 Most of these compounds have a high tendency to dissolve into water. Due to their widespread use, these monomers can easily leach into water and contaminate it, and their removal from water is not straight-forward. While other acrylates mentioned here are highly volatile and can be detected through techniques such as gas chromatography coupled with mass spectrometry (GC-MS), AM is even more challenging to detect and sequester. AM has a low vapour pressure and is not amenable to detection by GC-MS. Researchers have developed specific mass spectroscopy methods such as gas chromatography with electron capture detector (GC-ECD) and high-performance liquid chromatography with UV (HPLC-UV) to detect AM,7–10 although detailed pre-treatment of contaminated samples is required. For example, bromination of AM affords 1,2-dibromopropionamide that is unstable in water but has a high partition coefficient in organic solvents, and can be detected by the GC-ECD method. Additional extraction methods and detection techniques have been developed to enhance the limit of detection. In another protocol, AM was reacted with diazoalkane to produce a strong yellow-coloured product that could be detected through colorimetry. However, this colorimetric method works exclusively in an organic phase. All the above methods need chemical modification of AM prior to detection, and no suitable chemical method or reagent is available in the literature to detect AM in aqueous medium.
Here, we report two water-soluble pro-sensors (Ph-amn and Nap-amn) for direct detection of acryl compounds including AM in aqueous medium. We employed the S-to-N acyl transfer step, akin to the final step of native chemical ligation (Fig. 1a), to activate these pro-sensors.11–14 These pro-sensors were synthesized using a common scheme (Scheme S1, ESI†), and differ in the aryl unit attached to the amine end of L-alanine. While Ph-amn contains a phenylacetyl unit, Nap-amn contains a 1-naphtheleneacetyl residue protecting the amine end of L-alanine. We have been investigating the self-assembly of amino acid derivatives containing such rotationally flexible aromatic N-protecting residues, and have found that these derivatives respond to chemical and physical cues.15,16 In the current design, the carboxylic acid end of alanine was converted to thioester using 2-aminoethane thiol. A generalized structure of the compounds can be seen in Fig. 1b. Due to the terminal ammonium residue, these pro-sensors were freely water soluble. Upon an increase of pH, these compounds spontaneously undergo S-to-N acyl shift via a five membered transition state to yield the active sensor containing thiolate functionality, as indicated in Fig. 1b.
 | ||
| Fig. 1 (a) S-to-N acyl shift in the final step of intein mediated protein splicing. (b) Generalized chemical structure of prosensors Aryl-amn (Ph-amn and Nap-amn) that undergo similar S-to-N acyl shift upon pH increase to form active thiolate sensors (Ph-S and Nap-S). | ||
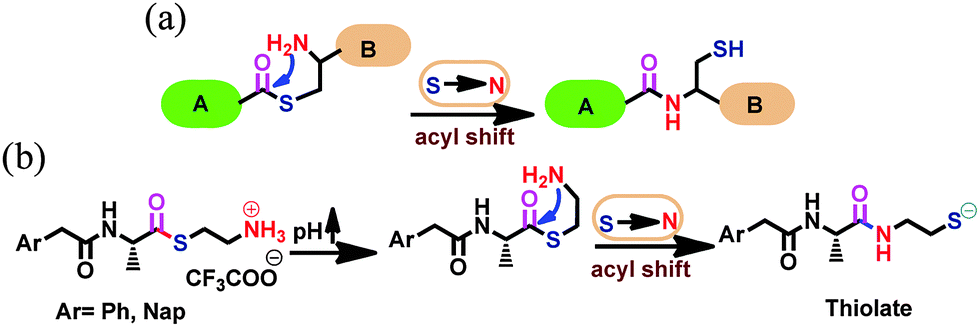
This transformation was characterized by 1H NMR spectroscopy in D2O for the Ph-amn pro-sensor (Fig. 2a). An increase of pH (using 1 M phosphate buffer of pD = 8) resulted in the instantaneous formation of Ph-S. During the whole process, the aqueous medium remained clear (as can be seen from the low scattering for blue data points in Fig. 2b). In contrast, when Ph-S synthesized independently according to Scheme S3, ESI,† was employed, it required strong heating to get solubilized in water, and then also, started precipitating gradually as the hot aqueous solution cooled. We believe that this is due to extensive intermolecular H-bonding between Ph-S molecules in the solid state, which needs to be overcome by water to solubilize it. Thus, use of a pro-sensor seemed to be a more straight-forward approach to yield clear solutions of Ph-S for further use.
 | ||
| Fig. 2 (a) 1H (400 MHz) NMR spectra in D2O of Ph-amn (10 mg mL−1), and of the product (Ph-S) formed upon increasing solution pH using 1 M phosphate buffer (pD = 8). (b) Results of DTNB assay (green symbols) and turbidity measurement (blue symbols) done at different time points after an increase of pH of solution containing Ph-amn (4 mg mL−1). The inset shows the initial phase of this reaction. H2O2 was added to the sample at 635 min. This time point is indicated by arrow. (c) SEM image of the helical tape-like assemblies present in the precipitate obtained upon addition of H2O2 to Ph-S. | ||
We investigated the pH-induced spontaneous transformation of Ph-amn to Ph-S further. 1H NMR experiments demonstrated rapid and exclusive intra-molecular S-to-N acyl shift of Ph-amn to form Ph-S (Fig. 2a). The formation of Ph-S under these conditions was complete within a couple of minutes. We also performed competitive studies using externally-added 1-aminohexane (HA) as a secondary nucleophile that can attack the thioester Ph-amn inter-molecularly (Fig. S1, ESI†). Irrespective of whether the pH increase was undertaken before or after the addition of HA, there was no evidence of any intermolecular attack of HA on Ph-amn. The formation and stability of Ph-S were also probed by 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) assay (Fig. 2b). As can be seen in the inset of Fig. 2b, the free thiolate concentration increased very rapidly after pH increase, peaking within about 15 min, before reaching a somewhat lower equilibrium concentration within about 20 min. The minor decrease in free thiolate concentration is presumably due to some aerial oxidation of the thiolate under the basic conditions. However, beyond this point, the thiolate concentration did not change for >10 h unless an external oxidant (such as H2O2) was added to the reaction mixture. Addition of H2O2 caused an immediate decrease of absorbance at 412 nm (Fig. 2b), due to conversion of Ph-S to its disulphide form. We also measured the turbidity of Ph-S solution during the whole process. Before H2O2 addition, the solution of Ph-S had negligible scattering and the sample was visually clear. Addition of H2O2 resulted in immediate formation of a copious white precipitate, which was reflected in a sharp increase in the turbidity (Fig. 2b). We confirmed by 1H NMR and mass spectrometry (Fig. S2 and S3, ESI†) that this enhanced turbidity was due to the formation of water-insoluble disulfide (Ph-SS-Ph). Scanning electron microscopy (SEM) observation of the precipitate showed the presence of tape like assemblies having a distinct right handed helicity (Fig. 2c), reflecting the chirality present in the molecule.
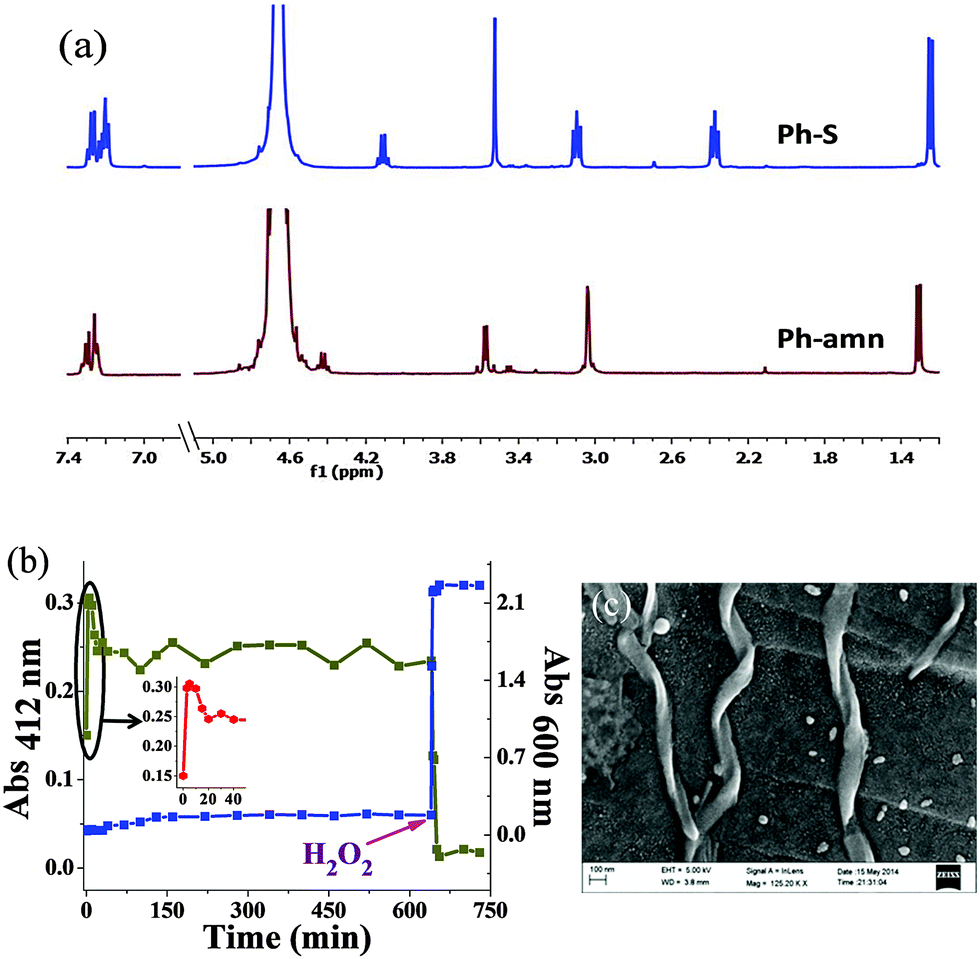
The above experiment indicated that the thiolate generated upon pH-induced S-to-N acyl transfer was kinetically stable at least for a few hours. We thus exploited the well-known Michael addition of thiolates with Michael acceptors like α,β-unsaturated carbonyl compounds.17,18 Here, we employed Ph-S to detect water soluble acryl compounds such as AM, AN, MA, EA and MM through the formation of Michael adducts with them. Upon addition of one equivalent of any of these compounds to the Ph-S solution, we noticed significant precipitation within a short interval (Fig. 3a). For all the acrylates (except MM), the precipitate formation saturated within 10 min. With MM, it took somewhat longer (ca. 20 min) to observe the emergence of the complete turbidity. This may be due to slower kinetics of reaction between MM and Ph-S. Turbidity measurements showed significantly high scattering values for the samples containing acrylates (Fig. 3b). We confirmed that this screening worked even for AM solutions prepared in tap water and also in simulated body fluid (SBF, Fig. 3c), and all test samples were highly turbid while all the control samples had very low turbidity. SBF has pH and ion content similar to that found in human serum plasma. It was used as a surrogate of protein-free biological fluid to exemplify the utility of Ph-S in detecting AM under biorelevant conditions. We conducted SEM imaging of the precipitates formed from the reaction between the acryl compounds and Ph-S. While Michael adducts of Ph-S and AM formed tape-like nano-assemblies (Fig. 3d), AN yielded thin fibres, MA yielded tapered cuboids (Fig. S4, ESI†). Under similar conditions, EA produced flat plate-like assemblies with Ph-S while MM produced bulky unstructured assemblies. The formation of all the Michael adducts was confirmed by mass spectrometry too (Fig. S5–S9, ESI†). With Ph-S, we observed considerable precipitation only at AM concentrations of ≥14 mM. At 14 mM AM concentration, the sample needed to be incubated for about 45 min to obtain the maximum turbidity (Fig. S10, ESI†).
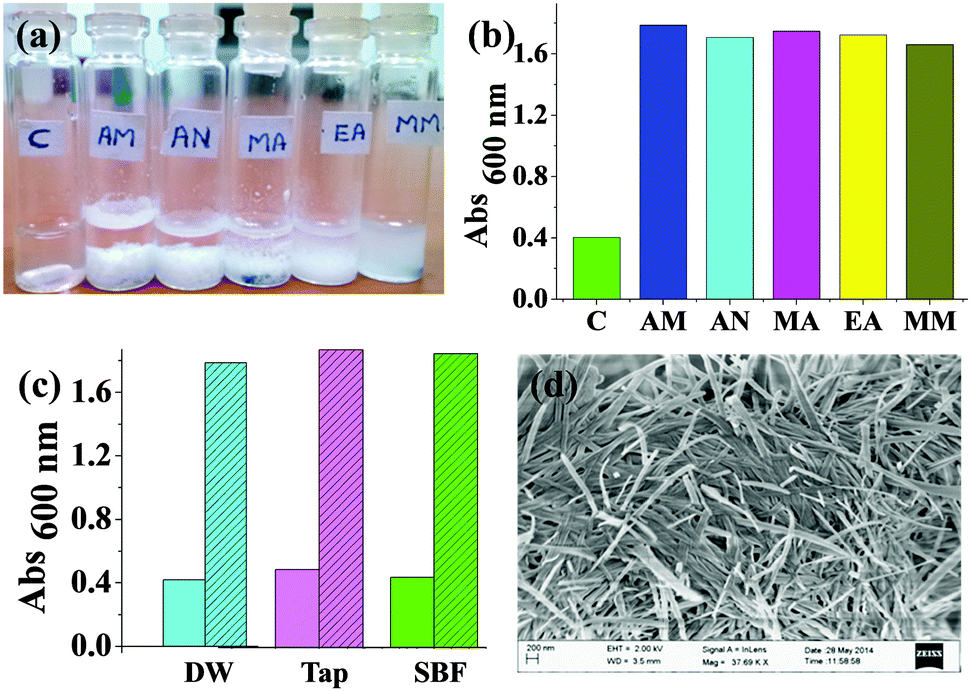 | ||
| Fig. 3 (a) Digital image of vials containing Ph-S (64 mM in 1 M carbonate buffer of pH = 9) to which different acrylates were added. C: control, AM: acrylamide, AN: acrylonitrile, MA: methyl acrylate, EA: ethyl acrylate and MM: methyl methacrylate. (b) Turbidity profiles of these samples. (c) Turbidity profiles of control (plain bars) and test (shaded bars) samples prepared in distilled water (DW), tap water and SBF. (d) SEM image of the precipitate obtained upon addition of AM to Ph-S in DW. | ||
Ph-S thus has a rather poor detection limit (14 mM) for AM. To enhance the detection limit, we employed Nap-amn that too undergoes rapid intramolecular S-to-N acyl shift upon pH increase (see Fig. S11, ESI,† for 1H NMR of Nap-S). Due to the presence of the naphthalene residue in this compound, we could employ fluorescence spectroscopy to detect AM using Nap-S. The Nap-S compound also undergoes analogous Michael reaction with AM to yield the adduct shown in Scheme S2, ESI.† To Nap-S solution we added different concentrations of AM. Generally, AM acts as a fluorescence quencher. However, in the present case, the fluorescence intensity of Nap-S increased with incremental addition of AM (Fig. 4a), while the control sample (to which only water was added) did not show any change. The fluorescence enhancement in test samples is attributed to the formation of the Nap-S + AM adduct (Fig. S12, ESI†) that exhibits stronger fluorescence compared to Nap-S. The enhancement of fluorescence was discernible even at AM concentrations as low as 2.5 ppm. For the 16 ppm of AM, an incubation time of approximately 30 min was needed to obtain complete enhancement of fluorescence (Fig. 4c). We observed minor, batch-to-batch differences in the fluorescence profiles of Nap-S originating from the source of the 1-naphthalene acetic acid precursor. However, this did not alter the ability to detect AM by an increase of fluorescence intensity. The powder X-Ray diffraction (PXRD) studies indicated that the Nap-S + AM adduct is highly amorphous and a broad peak was observed at ca. 23° (Fig. S13, ESI†). This corresponds to a d-spacing of 3.6 Å, indicating that the sample has pi-stacked naphthalene units. No further information could be extracted from the PXRD due to the amorphous nature of the sample.
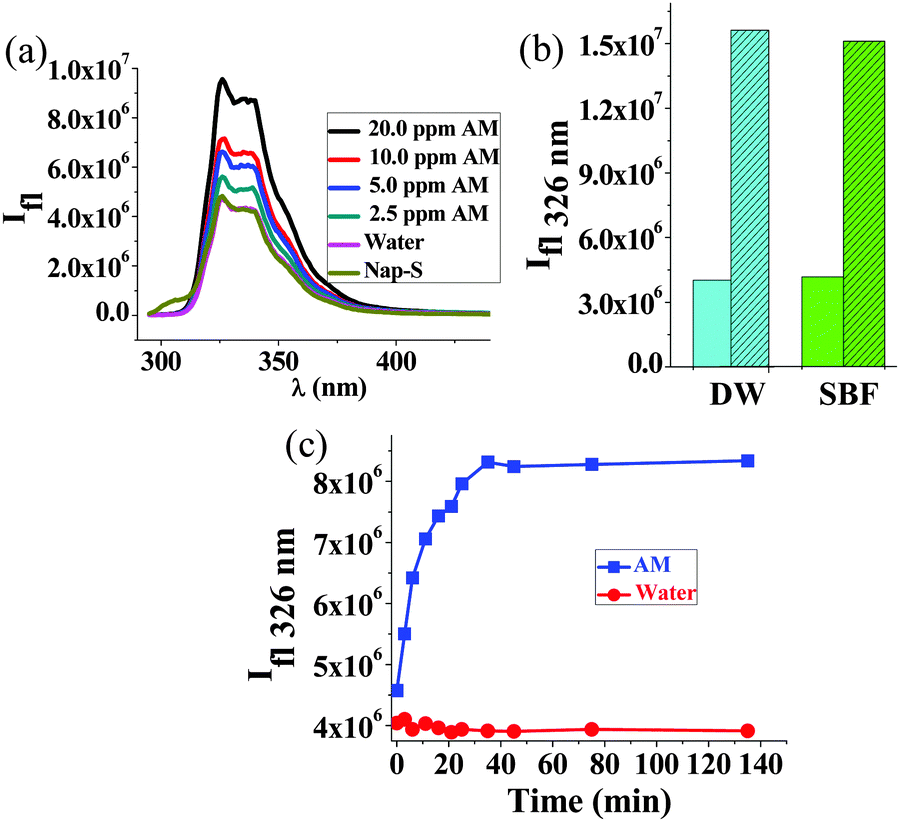 | ||
| Fig. 4 (a) Changes in fluorescence intensity of Nap-S solution (1 mM in distilled water) to which incremental amounts of AM was added. (b) Enhancement of fluorescence intensity at 326 nm upon addition of AM (30 ppm) in DW and SBF (plain bars: control samples, filled bars: samples containing AM). (c) Time profile of an increase in fluorescence intensity upon addition of AM (final concentration 16 ppm, blue symbols) to Nap-S (1 mM), compared to the control sample (red symbols) without AM. | ||
In conclusion, aryl-thioester pro-sensors (Ph-amn and Nap-amn) for detecting water soluble acrylates were synthesized. These compounds were activated through a spontaneous S-to-N acyl shift that occurs upon pH increase. The resulting thiolates provided easy and rapid indication of different water soluble acrylates. With AM, either tape-like nanoassemblies were obtained (upon employing Ph-S), or enhancement of fluorescence was noticed (with Nap-S). Using Nap-S, acrylamide could be detected at concentrations ≥2.5 ppm in the aqueous medium.
We acknowledge IISER Bhopal for the intramural funding. ARM also thanks IISER Bhopal for the institute fellowship. We acknowledge Mr Somnath Bhagat for independent synthesis and confirmation of the results reported.
Notes and references
- V. A. Myagchenkov and V. F. Kurenkov, Polym. -Plast. Technol. Eng., 1991, 30, 109–135 CrossRef CAS PubMed.
- M. R. Jain, C. Jain and R. C. Jain, J. Appl. Polym. Sci., 2013, 129, 2536–2543 CrossRef CAS PubMed.
- E. Tareke, P. Rydberg, P. Karlsson, S. Eriksson and M. Törnqvist, J. Agric. Food Chem., 2002, 50, 4998–5006 CrossRef CAS PubMed.
- J. G. F. Hogervorst, B. J. Baars, L. J. Schouten, E. J. M. Konings, R. A. Goldbohm and P. A. van den Brandt, Crit. Rev. Toxicol., 2010, 40, 485–512 CrossRef CAS PubMed.
- A. Shipp, G. Lawrence, R. Gentry, T. McDonald, H. Bartow, J. Bounds, N. Macdonald, H. Clewell, B. Allen and C. Van Landingham, Crit. Rev. Toxicol., 2006, 36, 481–608 CrossRef CAS PubMed.
- J. M. Rice, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2005, 580, 3–20 CrossRef CAS PubMed.
- V. V. Bessonov, A. D. Malinkin, O. I. Perederiaev, M. N. Bogachuk, S. V. Volkovich and V. Medvedev Iu, Voprosy pitaniia, 2011, 80, 79–83 CAS.
- Z. M. Geng, P. Wang and A. M. Liu, J. Chromatogr. Sci., 2011, 49, 818–824 CAS.
- J. Oracz, E. Nebesny and D. Zyzelewicz, Talanta, 2011, 86, 23–34 CrossRef CAS PubMed.
- C. G. Daughlon, Quantitation of acrylamide (and polyacrylamide): critical review of methods for trace determination/formulation analysis & future-research recommendations, The California Public Health Foundation, Final Report No. CGD-02/88, June 1988.
- O. Bol'shakov, J. Kovacs, M. Chahar, K. Ha, L. Khelashvili and A. R. Katritzky, J. Pept. Sci., 2012, 18, 704–709 CrossRef PubMed.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS.
- S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- J. S. Zheng, S. Tang, Y. C. Huang and L. Liu, Acc. Chem. Res., 2013, 46, 2475–2484 CrossRef CAS PubMed.
- A. Reddy M and A. Srivastava, Soft Matter, 2014, 10, 4863–4868 RSC.
- P. S. Yavvari, A. Reddy M and A. Srivastava, RSC Adv., 2013, 3, 17244–17253 RSC.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. X. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc02721g |
| This journal is © The Royal Society of Chemistry 2015 |