
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
G-quadruplex ligands exhibit differential G-tetrad selectivity†
D. D.
Le
a,
M.
Di Antonio
ab,
L. K. M.
Chan
a and
S.
Balasubramanian
*abc
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
bCambridge Research UK, Cambridge Institute, Li Ka Shing Centre, Robinson Way, Cambridge, CB2 0RE, UK
cSchool of Clinical Medicine, University of Cambridge, Addenbrooke's Hospital, Hills Road, Cambridge, CB2 0SP, UK. E-mail: sb10031@cam.ac.uk
First published on 3rd April 2015
Abstract
A rapid and simple equilibrium-binding assay mediated by ligand-induced fluorescence quenching of fluorophore-labelled G-quadruplex (G4) structures enabled quantitative interrogation of mutually exclusive ligand binding interactions at opposed G-tetrads. This technique revealed that the ligands TmPyP4, PhenDC3, and PDS have differential chemotype-specific binding preferences for individual G-tetrads of a model genomic G4 structure.
While the primary sequences of DNA and RNA encode the fundamental information necessary for cellular function, it is the secondary structure adopted by these molecules that, in part, dynamically regulates their activity.1 One such regulatory structure is the guanine quadruplex, which has been shown to influence core cellular processes such as replication, transcription and translation.1,2 Structures of this type arise in G-rich sequences where guanines assemble into multi-layered tetrad planes (G-tetrads), stabilized by Hoogsteen hydrogen-bonding and π–π stacking (Fig. 1). Importantly, G4 structures may be associated with DNA instability and cancer progression; thus, pharmacological targeting of G4s has considerable potential for probing cancer biology and modulating cancer phenotypes.3
 | ||
| Fig. 1 Tetrad-specific fluorescence quench equilibrium dissociation binding assay for G-quadruplex ligands. | ||
The disparate features of G4 architecture, revealed by structural studies, present unique topologies that determine the potential modes of ligand binding: tetrad-stacking, groove-binding, and loop-binding.4 Furthermore, individual G-tetrad ends of a given G4 structure are chemically distinct environments that influence small molecule interactions. Given this structural heterogeneity, the development of selective G4 ligands has, to some extent, been constrained by the resolution of current techniques to quantitate equilibrium ligand binding at specific sites within G-quadruplexes. Because the most ubiquitous G4 ligand chemotypes are based on planar aromatic scaffolds, which interact primarily via π–π stacking to G-tetrad ends, these compounds are predicted to access both G-tetrads of a given G4 structure.5 However, widely adopted methods to characterize G4 ligand interactions, such as G4 thermal shift (G4 FRET-melting) and G4 fluorescent intercalator displacement (G4-FID)—while rapid and simple to implement—can not quantitate the equilibrium constants of individual G-tetrad binding sites.6 To better probe the binding characteristics of G-quadruplexes and their small molecule ligands, described here is a fluorescence-based binding assay that rapidly, simply, and accurately measures apparent equilibrium dissociation constants (KappD) at defined sites on G4 structures.
Building upon the utility of reported fluorescence quench assays, the present work describes differentially fluorophore-labelled G4-forming oligonucleotides (oligos), which exhibit quenching mediated by proximal ligand binding at individual G-tetrads (Fig. 1).7 This ligand-induced phenomenon was developed into a general G4 binding assay to enable measurement of KappD for the established ligands TmPyP4, PhenDC3, and pyridostatin (PDS) with several G4 structures.8 The assay platform was based on initial observations that the G4-specific ligand PDS causes dose-dependent loss of fluorescence emission on a 5′-Cy5 labelled DNA oligo derived from the human telomeric repeat sequence (5′Cy5_hTelo) (Fig. 2A). The fluorescence quenching phenomenon was shown to require a folded G4 structural context, as the ligand-induced effect was abrogated by nuclease digestion and incomplete G-quadruplex folding in Li+ conditions (Fig. 2B; Fig. S1, S2A and B, ESI†).9 Importantly, quenching was annulled by competition with an unlabelled hTelo oligo, indicating that PDS interacts specifically and reversibly with the labelled G4 structure to suppress fluorescence emission; whereas, competition with a non-G4 mutant oligo did not reverse fluorescence quenching (Fig. 2C). The competition results corroborate direct quenching measurements, despite fluorophore-induced structural polymorphism common to hTelo constructs (Fig. 2B and C and Fig. S2C, ESI†). Taken together, these data support a proximal quenching mechanism whereby ligands bind to structured G4 elements in the vicinity of an excited-state fluorophore to induce non-radiative dissipation. Thus, the extent of fluorescence quenching is indicative of ligand binding.
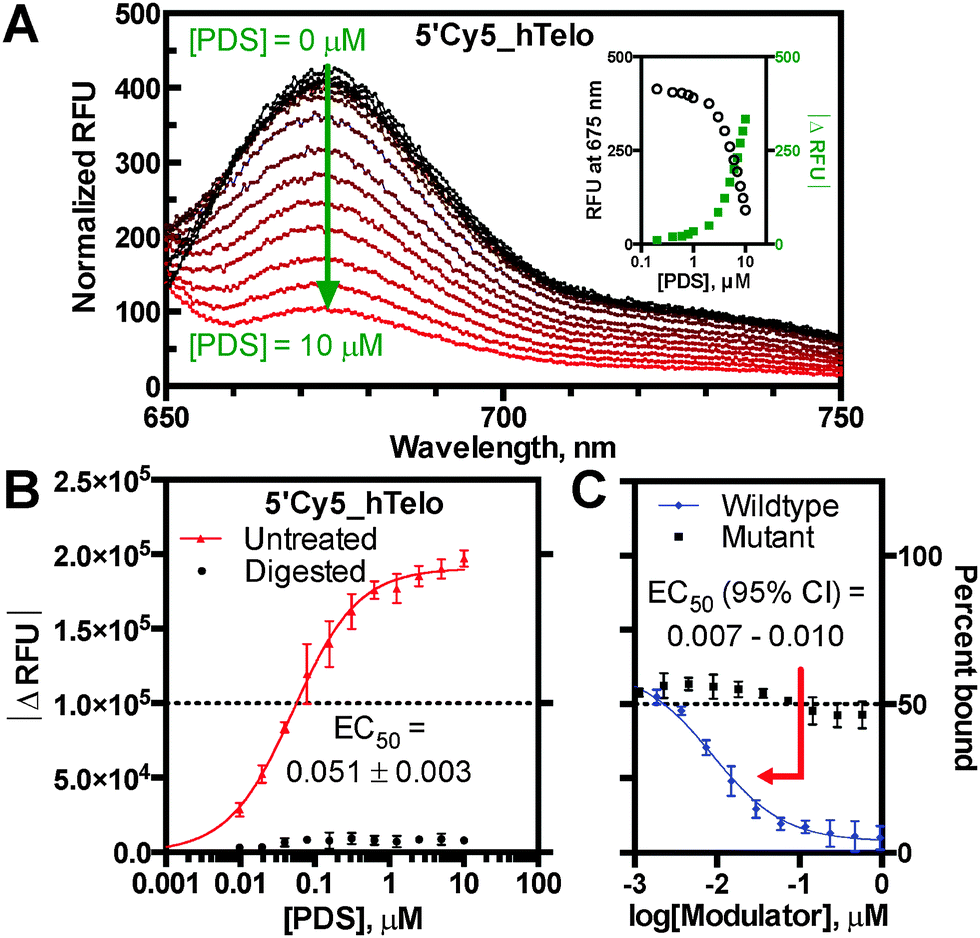 | ||
| Fig. 2 PDS binding to human telomeric DNA G4. (A) Dose-responsive Cy5 emission spectrum (inset: absolute change in RFU), (B) PDS-induced saturation of fluorescence response at 680 nm and nuclease digestion control, (C) competition binding assay. | ||
To cross-validate the present assay results with previously reported equilibrium dissociation constants, saturation binding analysis was performed with the widely used G4 ligand TmPyP4 on several described G4 structures: 5′-untranslated region (5′UTR) sequence of the NRAS proto-oncogene RNA transcript, c-Myc and c-kit proto-oncogene promoters, and the human telomeric repeat sequence (Table S2 and Fig. S3, ESI†).7c,10 Initially, comparisons of measured values to reported equilibrium constants were complicated by claims of multiple binding events (Table S2, ESI†). Instances of two discrete binding transitions were described: a high-affinity event and one of low-affinity. It was reasoned that the transitions represent individual TmPyP4 binding events at G-tetrad ends of a G4 structure with unique local topologies that cause disparate binding equilibria. Therefore, to probe ligand binding at either end of a G4 structure, differentially Cy5 end-labelled oligos were used to measure 5′-tetrad versus 3′-tetrad quenching caused by TmPyP4 interaction (Fig. 3A). Analysis of the hTelo sequence revealed KappD = 1.9 ± 0.2 μM and KappD = 0.15 ± 0.01 μM for the 5′Cy5_hTelo and 3′Cy5_hTelo constructs, respectively (Fig. S3D, ESI†). These measurements are consistent with reported dissociation equilibria determined by isothermal titration calorimetry describing multiple binding events for TmPyP4 binding to hTelo: 2.0 ± 0.2 μM and 0.25 ± 0.3 μM (Table S2 and Fig. S4, ESI†).11 Similarly, high-affinity and low-affinity apparent binding constants were observed for TmPyP4 with both cKit1 and cMyc structures, in accordance with literature values (Fig. 3B, Fig. S3 and S4, ESI†). Corroborating these observations are structural studies that have suggested intrinsic ligand binding preferences for a particular G-tetrad.12 For example, NMR spectroscopic analysis of the c-Myc G-quadruplex in complex with TmPyP4 indicates preferential binding to the 5′-end surface.12 Importantly, measured tetrad-specific equilibrium dissociation results, enabled by the fluorescence quench assay, are consistent with the proposed binding model: 5′Cy5_cMyc KappD = 0.015 ± 0.003 μM (high-affinity) and 3′Cy5_cMyc KappD = 0.31 ± 0.02 μM (low-affinity) (Fig. S3B and C, ESI†). Taken together, the attribution of G-tetrad selectivity to reported TmPyP4 binding equilibria builds upon a general structural rationale for the observations of dual binding events.
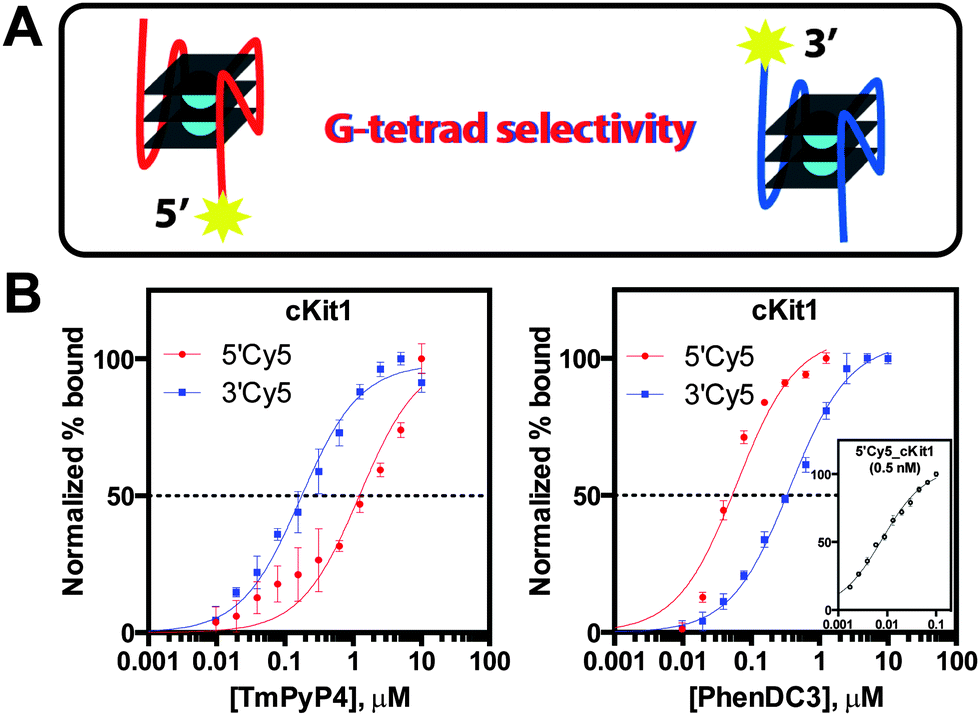 | ||
| Fig. 3 G-tetrad selectivity analysis. (A) 5′-tetrad versus 3′-tetrad ligand binding, (B) inverse G-tetrad selectivity between TmPyP4 and PhenDC3 with cKit1 (inset: [5′Cy5_cKit1] = 0.5 nM). | ||
To better understand the binding preferences of common G4 ligands, the present assay was used to quantitate G-tetrad selectivity of TmPyP4, PhenDC3, and PDS toward the model G4 structure cKit1. Analysis of differentially Cy5 end-labelled cKit1 revealed that TmPyP4 exhibits an approximate 8-fold selectivity for the 3′-end tetrad (Fig. 3). In contrast, PhenDC3 displayed the reverse selectivity trend, demonstrating 46-fold tighter 5′-tetrad binding (Fig. 3). These results highlight the differences in chemical environment between G-tetrads of a G4 structure that determine ligand-specific binding preferences. Unlike TmPyP4 and PhenDC3, the ligand PDS exhibited nearly equivalent binding affinity toward both G-tetrads of the cKit1 structure (Fig. S5, ESI†). Quantitation of ligand equilibrium dissociation constants revealed the inherent binding preferences of each chemotype toward the individual G-tetrad ends of the cKit1 structure. Such analyses provide a higher degree of structural resolution to dissect the molecular determinants of ligand binding at G-tetrads. Moreover, the unprecedented ability to rapidly and simply probe G-tetrad specific binding events is expected to advance G4 ligand design toward higher selectivity and potentially novel tetrad-specific pharmacological activity.
Ligand-induced fluorescence quenching of labelled G4-forming oligonucleotides has been developed into a rapid and simple equilibrium-binding assay. Cross-validation to literature values describing ligand interactions with several important G4 structures indicates that the present method produces accurate dissociation constants. A key feature of this fluorescence quench assay is the ability to distinguish KappD derived from defined G-tetrads, enabling targeted structure–activity studies aimed at improving ligand design and to probe tetrad-specific G4 topology. It was demonstrated that G4 ligands exhibit strong and varied G-tetrad preferences for genomic G4 structures. This method facilitates a rethinking of G4 ligand binding and selectivity: rather than being simply sequence-specific (e.g. cKit1 vs. cMyc), G4 ligands bind to a given G-quadruplex structure at distinct G-tetrads with differential binding affinities.
This work was supported by the following grants: Cancer Research UK Programme, BBSRC BB/K018043/1 and EPSRC EP/K039520/1. We thank Dr Chris Lowe for his constructive comments and for proofreading the manuscript.
Notes and references
- (a) M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef PubMed; (b) R. Rodriguez and K. M. Miller, Nat. Rev. Genet., 2014, 15, 783–796 CrossRef PubMed.
- (a) P. Murat and S. Balasubramanian, Curr. Opin. Genet. Dev., 2014, 25, 22–29 CrossRef CAS PubMed; (b) A. Bugaut and S. Balasubramanian, Nucleic Acids Res., 2012, 40, 4727–4741 CrossRef PubMed.
- (a) A. L. Wolfe, K. Singh, Y. Zhong, P. Drewe, V. K. Rajasekhar, V. R. Sanghvi, K. J. Mavrakis, M. Jiang, J. E. Roderick, J. Van der Meulen, J. H. Schatz, C. M. Rodrigo, C. Zhao, P. Rondou, E. de Stanchina, J. Teruya-Feldstein, M. A. Kelliher, F. Speleman, J. A. Porco Jr., J. Pelletier, G. Rätsch and H. G. Wendel, Nature, 2014, 513, 65–70 CrossRef CAS PubMed; (b) G. Biffi, D. Tannahill, J. Miller, W. J. Howat and S. Balasubramanian, PLoS One, 2014, 17, e102711 Search PubMed; (c) T. Shalaby, G. Fiaschetti, K. Nagasawa, K. Shin-ya, M. Baumgartner and M. Grotzer, Molecules, 2013, 18, 12500–12537 CrossRef CAS PubMed; (d) S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef PubMed.
- (a) S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef PubMed; (b) T. Kimura, K. Kawai, M. Fujitsuka and T. Majima, Tetrahedron, 2007, 63, 3585–3590 CrossRef PubMed.
- Q. Li, J. F. Xiang, Q. F. Yang, H. X. Sun, A. J. Guan and Y. L. Tang, Nucleic Acids Res., 2013, 41, D1115–D1123 CrossRef PubMed.
- (a) J. L. Mergny and J. C. Maurizot, ChemBioChem, 2001, 2, 124–132 CrossRef; (b) A. De Cian, L. Guittat, M. Kaiser, B. Saccà, S. Amrane, A. Bourdoncle, P. Alberti, M. P. Teulade-Fichou, L. Lacroix and J. L. Mergny, Methods, 2007, 42, 183–195 CrossRef PubMed; (c) E. Largy, F. Hamon and M. P. Teulade-Fichou, Anal. Bioanal. Chem., 2011, 400, 3419–3427 CrossRef PubMed; (d) P. L. Tran, E. Largy, F. Hamon, M. P. Teulade-Fichou and J. L. Mergny, Biochimie, 2011, 93, 1288–1296 CrossRef PubMed.
- (a) J. Alzeer, B. R. Vummidi, P. J. Roth and N. W. Luedtke, Angew. Chem., Int. Ed., 2009, 48, 9362–9365 CrossRef PubMed; (b) J. Alzeer and N. W. Luedtke, Biochemistry, 2010, 49, 4339–4348 CrossRef PubMed; (c) M. Faudale, S. Cogoi and L. E. Xodo, Chem. Commun., 2012, 48, 874–876 RSC.
- (a) E. Izbicka, R. T. Wheelhouse, E. Raymond, K. K. Davidson, R. A. Lawrence, D. Sun, B. E. Windle, L. H. Hurley and D. D. Von Hoff, Cancer Res., 1999, 59, 639–644 Search PubMed; (b) A. De Cian, E. Delemos, J. L. Mergny, M. P. Teulade-Fichou and D. Monchaud, J. Am. Chem. Soc., 2007, 129, 1856–1857 CrossRef PubMed; (c) R. Rodriguez, S. Müller, J. A. Yeoman, C. Trentesaux, J. F. Riou and J. S. Balasubramanian, J. Am. Chem. Soc., 2008, 130, 15758–15759 CrossRef PubMed.
- (a) C. C. Hardin, E. Henderson, T. Watson and J. K. Prosser, Biochemistry, 1991, 30, 4460–4472 CrossRef; (b) J. R. Williamson, M. K. Raghuraman and T. R. Cech, Cell, 1989, 59, 871–880 CrossRef.
- (a) A. Siddiqui-Jain, C. L. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593–11598 CrossRef PubMed; (b) J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2007, 35, 406–413 CrossRef PubMed; (c) K. N. Luu, A. T. Phan, V. Kuryavyi, L. Lacroix and P. J. Patel, J. Am. Chem. Soc., 2006, 128, 9963–9970 CrossRef PubMed.
- A. Arora and S. J. Maiti, J. Phys. Chem. B, 2008, 112, 8151–8159 CrossRef PubMed.
- (a) A. T. Phan, V. Kuryavy, H. Y. Gaw and D. J. Patel, Nat. Chem. Biol., 2005, 1, 167–173 CrossRef PubMed; (b) W. J. Chung, B. Heddi, F. Hamon, M. P. Teulade-Fichou and A. T. Phan, Angew. Chem., Int. Ed., 2014, 53, 999–1002 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed methods, supplemental figures and tables. See DOI: 10.1039/c5cc02252e |
| This journal is © The Royal Society of Chemistry 2015 |