
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-inspired CO2 conversion by iron sulfide catalysts under sustainable conditions†
A.
Roldan
a,
N.
Hollingsworth
a,
A.
Roffey
a,
H.-U.
Islam
ab,
J. B. M.
Goodall
a,
C. R. A.
Catlow
a,
J. A.
Darr
a,
W.
Bras
b,
G.
Sankar
a,
K. B.
Holt
a,
G.
Hogarth
a and
N. H.
de Leeuw
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: n.h.deleeuw@ucl.ac.uk
bEuropean Synchrotron Radiation Facility, BP220, Grenoble F38043, France
First published on 24th March 2015
Abstract
The mineral greigite presents similar surface structures to the active sites found in many modern-day enzymes. We show that particles of greigite can reduce CO2 under ambient conditions into chemicals such as methanol, formic, acetic and pyruvic acid. Our results also lend support to the Origin of Life theory on alkaline hydrothermal vents.
Hydrothermal vents consist of porous chimney structures composed of colloidal barriers of continually forming iron sulfides in three-dimensional cavities.1 The chimneys comprise mostly iron, nickel and cobalt sulfides as well as silica gel, ferrous hydroxide and oxyhydroxides.2 It has been proposed that at the interface between hydrothermal fluids and the primordial ocean, H2 and CO2 would have reacted together to form small organic molecules, catalyzed by the FeS membranes formed in the plumes of the vents.1d,3 Hydrothermal vents found on the ocean floor can be divided into two main types, those with acidic and alkaline effluents. Acidic hydrothermal vents, termed ‘black smokers’ are located directly above magma chambers,4 with a hot (up to 405 °C) acidic (pH 2–3) effluent.5 In contrast, alkaline hydrothermal vents have cooler (40–91 °C), more basic effluent (pH 9–11).6 Furthermore, the cavities in the chimneys restrict the diffusion of the vent springs leading to a natural chemi-osmotic potential owing to the contrast in pH,3 which could provide the driving force required to overcome the initial thermodynamically unfavorable CO2 reduction step.7
An attractive suggestion is the hypothesis that iron-sulfide minerals, such as greigite (Fe3S4), found in the chimney cavities of hydrothermal vents,2 catalyzed CO2 reduction, forming a primitive acetyl-CoA pathway similar to that in contemporary enzymes.8 In fact, greigite is structurally similar to the Fe4S4 clusters found in ferredoxins,9Fig. 1A, which have been shown to act as electron-transfer sites and to be catalytically active centers for molecule transformations.10 Such enzymes are highly product-specific and efficient, shown for example in formate dehydrogenases, which are able to reduce CO2 to formate under moderate conditions, i.e. at low temperatures and pressures and at neutral pH.11 A recent study has highlighted the catalytic nature of greigite, showing that CO2 could be converted to CH4 and CO, but no solution-based products were detected in that study – an essential requirement for prebiotic chemistry.12 In addition, FeS has been shown to catalyze CO2 reduction13 and into a range of thiols in the presence of H2S.14
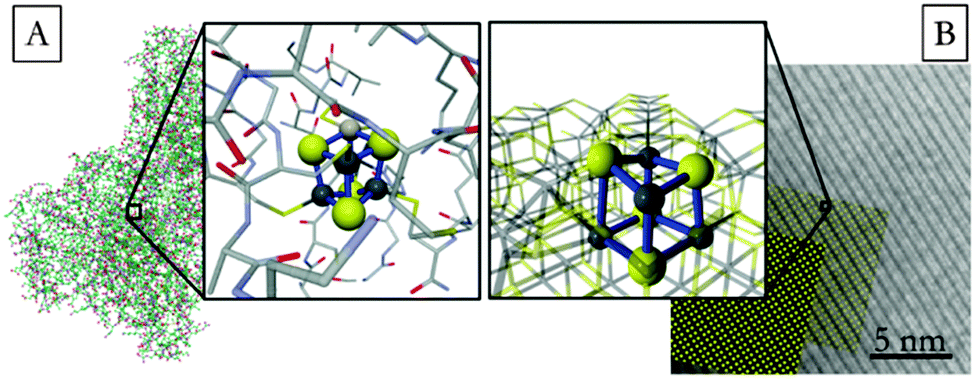 | ||
| Fig. 1 Schematic representation of (A) the ferredoxin center of the CO dehydrogenase enzyme,11f (B) the greigite surface, Fe3S4(001), showing enhanced the cubane structure. | ||
In this study, we show that CO2 can be reduced to a range of small organic solution-based molecules that are required for the commencement of pre-biotic chemistry. The use of a greigite surface allows such transformations at a low overpotential, atmospheric pressure and room temperature. Furthermore, we have elucidated the mechanism of formation of formic acid and methanol, which explains the requirement of an alkaline environment.
Highly faceted greigite nanoparticles have been synthesized through the decomposition of iron(III) dithiocarbamate. Metal dithiocarbamates have been shown to be excellent precursors to metal sulfide materials.15 The particle morphology is a rectangular plate with faces terminating at (001) and edges at (111) surfaces (see ESI†). Computer modelling of both bulk materials and the surfaces predicts bond lengths that closely resemble those found in the synthesized particles, with differences of only 0.06 ± 0.02 Å and 0.13 ± 0.02 Å for the short and long bonds.16 These are also very similar to the Fe–S distances found in the CO dehydrogenase enzyme, Δd(Fe–S) = 0.01 ± 0.02 Å and 0.08 ± 0.02 Å.11f The slightly longer Fe–S distances found in synthetic and modeled greigite, compared with the enzymatic cubane cluster, are due to periodic packing of the Fe and S atoms in a crystal lattice, which is not applicable in the enzyme.
A series of electrochemical reduction experiments were performed at room temperature and pressure, using a greigite-modified electrode in CO2-saturated aqueous solutions at pH 4.5, 6.5 and 10.5. The nano-carbon dispersed greigite particles were drop-coated onto a carbon rod electrode and the electrode potential was cycled continuously between 0.2 and −0.8 V vs. NHE at 1 mV s−1. Dissolved reduction products were detected ex situ at periodic intervals using quantitative 1H-NMR analysis. The major reaction product under all three pH-conditions was formic acid and the quantity detected with time is shown in Fig. 2A. Substantially more formic acid is generated at pH 6.5 than at pH 10.5; only a small quantity is produced at pH 4.5. Constant potential electrolysis experiments at pH 6.5 showed the onset of formic acid production at 0.4 V vs. NHE (see ESI†).
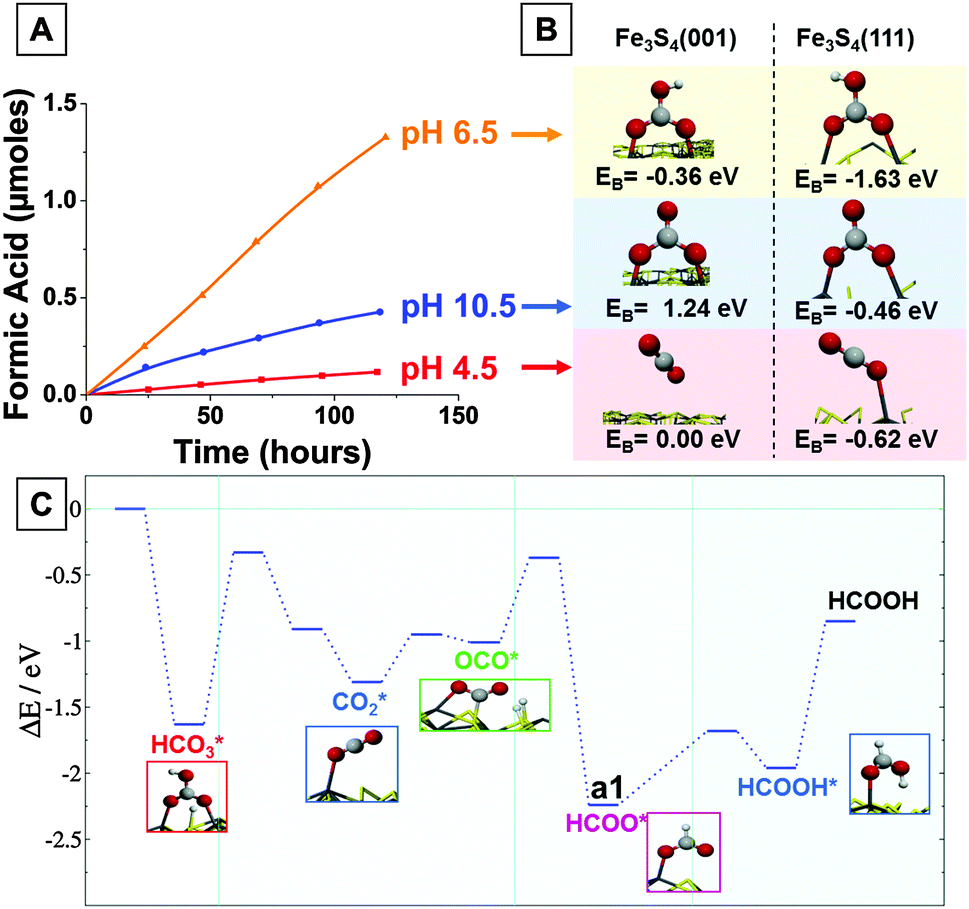 | ||
| Fig. 2 (A) Formic acid formation as a function of time under different pH conditions; (B) representation of reactants on both the (001) and (111) surfaces as a function of the solution pH, binding energies (EB) provided as inset; (C) potential energy surface for the mechanism of HCO3− reduction to HCOOH on the Fe3S4(111). Adsorbed intermediate species are denoted by * and their proposed structures are shown as insets in the figure. | ||
The computer simulations, using accurate ab initio techniques based on the Density Functional Theory (see ESI† for details), suggest that the disparity in formic acid production over the pH range can be attributed to the type and concentrations of aqueous species present in the CO2-saturated solutions. In agreement with the equilibrium constants: at low pH, dissolved CO2 mainly exists as a neutral molecule; at neutral pH, the dominant solution species is HCO3− (bicarbonate) and at high pH, CO32− (carbonate) is the majority species. These species interact with either the (001) or the (111) surfaces of the greigite particles. The calculations reveal that on the (001) surface the CO2 molecule experiences electrostatic repulsion between anionic surface sulfur atoms and the lone pairs of the molecule's oxygen atoms and it therefore does not bind to this surface (Fig. 2B). In contrast, the metallic centers on the (111) surface bind CO2 through an Fe–O bond with a binding energy (EB) of −0.62 eV. However, the molecule remains linear and not activated.17 Thus, at pH 4.5 the major solution species, i.e. neutral CO2, either does not bind to the surface or if it binds, it is not activated for further reaction, although the over-potential and elevated pressure existing at the hydrothermal vents may well enhance the interaction of gases on a substrate beyond that shown in our calculations. At pH 6.5, the major species present in solution is HCO3−, which binds to both the (001) and (111) surfaces through two O–Fe bonds perpendicular to the surface, thereby releasing energies of 0.36 eV and 1.63 eV, respectively. In basic solution (pH 10.5) the major species in solution is CO32−, which does not favorably adsorb onto the (001) surface (EB = +1.24 eV) but binds to the (111) surface with EB = −0.46 eV. Based on these values, the adsorption of HCO3− to the (111) and (001) surfaces and CO2 and CO32− to the (111) surface should be considered as steps in possible routes to products.
Clearly, water will compete with these species for binding at the metallic centers. The H2O molecule binds with EB = −0.42 eV on the (001) surface and considering that water is present in vast excess, competitive adsorption of HCO3− (EB = −0.36 eV) on the same surface is therefore unlikely, leaving only H2O bound to the (001) surface at all pH values considered. On the (111) surface, water binds with EB of −0.56 eV, this time out-competing the CO32− species (EB = −0.46 eV). As such, only HCO3− is able to adsorb on the (111) surface, owing to a much higher binding energy, by ΔEB = −1.07 eV, compared to H2O, implying that the (111) is the most probable surface for CO2 reduction and adsorbed HCO3− is the most likely reactive species. Consistent with the computed binding energies, the fastest experimentally observed rate of CO2 conversion to formic acid is observed at pH 6.5. Although other solution species dominate at pH 4.5 and 10.5, some HCO3− will still be present, allowing the conversion to formic acid to proceed under these conditions albeit at reduced rates. A further plausible explanation for the disparity of product concentrations, shown in Fig. 2A, are the competitive processes occurring at the different pHs. The simulations suggest that active sites deactivate at high pH due to surface OH accumulation, which adsorbs strongly on the Fe centres. This can be seen experimentally in the reduction in formic acid production over time at pH 10.5. At pH 6.5, the cycling potential bias of up to 0.2 V vs. NHE is enough to remove the hydroxyl groups. In addition, H2 generation from the reduction of adsorbed H at low pH (pH 4.5) competes with HCO3− reduction, thereby decreasing the reduction efficiency.
Based on the calculated binding energies, we propose a reaction mechanism to transform adsorbed HCO3− to formic acid on the Fe3S4(111) surface with surface H atoms, generated through dissociation of water. To identify the HCO3− transformation reactions, many intermediates and transition states were explored, leading to the multiple pathways plotted in Fig. S23 (ESI†). Our calculated energy barriers agree with the substantial kinetic barriers found in previous experiments on the formation of HCOO− and CO.18 However, the energy barriers are expected to decrease with applied negative electrode potential, and the model therefore represents the ‘worst case scenario’ for the energy barriers that need to be overcome in the conversion of HCO3− to formic acid.
Fig. 2C shows the computed thermodynamic and kinetic energy profile for the reduction of HCO3− on the (111) surface. As described above, adsorption of water onto the (111) surface releases 0.56 eV per molecule, although the energy barrier to dissociate the molecule to generate the surface H atoms and OH is calculated to be 0.94 eV. HCO3− binds favorably with the surface with EB = −1.63 eV to give HCO3*, which further reacts with a co-adsorbed H atoms to release H2O, requiring 1.31 eV to overcome the energy barrier. Although this is a substantial barrier, we postulate that if the system is not immediately equilibrated this barrier can be overcome by the energy released in the preceding adsorption step. An additional energy input of 0.36 eV is then required to activate CO2* to a bent geometry (OCO*), where both O and C interact with Fe and S surface atoms, respectively. This structure is stabilized by neighboring adsorbed H atoms, as shown in Fig. 2C inset. Hydrogenation of OCO* on the carbon atom results in intermediate a1 (HCOO*) and addition of a further H atom finally leads to formation of formic acid. Formic acid is bound to the surface (EB = −1.10 eV) and some energy is required to release the formic acid into solution.
The computed reaction profile shows that from the zero energy starting point the transformation of HCO3− into formic acid is thermodynamically and kinetically favorable. The reaction is driven by the energy released by thermodynamically favorable steps such as adsorption of the HCO3− and generation of the formate (a1) on the (111) surface. However, for the reaction to proceed, H ad-atoms are required on the surface, which requires an energy input of 0.94 eV from adsorbed molecular water. In our experiments, we have applied a modest over-potential of up to 1.1 eV, which is more than sufficient to generate the H ad-atoms required for this reaction. Thus, the calculations reveal a feasible mechanism for the production of formic acid under the moderate experimental conditions employed.
In addition to formic acid, other reaction products were experimentally detected under the three pH conditions investigated, as shown in Fig. 3A–C. At pH 4.5, low concentrations of acetic acid and methanol were detected, in accord with the low production of formic acid. On increasing the pH to 6.5, acetic acid, methanol and pyruvic acid were all detected in higher concentrations. At pH 10.5, the only additional product to formic acid is acetic acid, in intermediate concentrations. The concentrations of the products have been justified earlier, when we considered the concentration of HCO3− in solution. The disparity in the reduced products at varying pH can be accounted for by considering viable routes to methanol production. The overall faradaic efficiency for the production of formic, acetic, and pyruvic acid and methanol is calculated to be ca. 8% at pH 6.5.
 | ||
| Fig. 3 Formation of formic acid, acetic acid, methanol and pyruvic acid at (A) pH = 4.5; (B) pH = 6.5; (C) pH = 10.5. (D) Potential energy profile for the mechanism of HCO3− reduction to CH3OH, on the Fe3S4(111). | ||
Calculated reaction profiles reveal energetically feasible routes for methanol production, shown in Fig. 3D. The initial energetic profile is synonymous with that for formic acid production from HCO3− on the Fe3S4(111) surface (Fig. 2C), leading to the activated, bent OCO* intermediate. At this point, hydrogenation of the carbon atom eventually leads to formic acid, but an alternative route is hydrogenation of an oxygen atom to give intermediate b1 (COOH*). Although intermediate a1 is more stable thermodynamically than b1, by 1.1 eV, the difference between their transition states is only 0.1 eV, suggesting that both processes may take place competitively. Exothermic hydrogenation of the OH group of b1 leads to free H2O and CO* bound to Fe (b2). This intermediate is reduced to b3 (HCO*) after overcoming an energy barrier of 0.7 eV. At this stage, oxygen is preferentially hydrogenated, leading to b4 (HCOH*). Further hydrogenation of carbon leads to CH2OH, under a thermodynamic driving force (0.8 eV); although the transition state is 1.1 eV above the previous intermediate. Finally, a 1.2 eV barrier must be overcome for the final hydrogenation step to produce methanol (CH3OH*). Similar to the reaction pathway to form formic acid, the calculated reaction profiles suggest that methanol production is kinetically and thermodynamically favorable.
The formation of products such as acetic and pyruvic acid is explained by the slow process of releasing methanol into the solution, the activation barrier of b4 to b5 to methanol, as well as the endothermic nature of the final step. Consequently, b4 and b5 and methanol compounds accumulate on the surface where they can participate in coupling reactions to generate acetic or pyruvic acid. At low pH, however, a large amount of H is available on the surface, preventing significant coverage by b4 and b5, whereas at lower coverage of H ad-atoms (pH 10.5), the adsorbed CH2OH is more likely to undergo a dehydration reaction with formic acid to produce acetic acid. Formation of pyruvic acid at pH 6.5 is proposed to be the result from the dehydration of acetic acid and formic acid, which were present in higher concentrations at this pH. The relatively low concentrations of formic and acetic acid at pH 4.5 and 10.5 reduced the likelihood of pyruvic acid formation, which was hence not observed under these conditions.
This study has shown that the iron sulfide mineral greigite is able to promote the formation of small soluble organic molecules from CO2 in aqueous solution on application of a relatively small potential and under mild conditions. We found that pH 6.5 is the optimum pH for the effective reduction of dissolved CO2 at the greigite surface, with soluble organic molecules formed at an 8% faradaic efficiency. When we relate this pH to the acidic and alkaline hydrothermal vent systems on the ocean floor, only the alkaline hydrothermal vents provide the environment where this pH can be achieved, when considering their effluents at pH 10 mixing with the primordial ocean of pH 5.5.19 We note that a cycling potential is harder to justify in nature. However, Fe3S4 surfaces would be constantly regenerated due to its continual synthesis from the vent systems, and fresh Fe3S4 surfaces would thus be available continuously without cycling.13 The electro-potentials required are plausibly derived from galvanic interactions between mineral surfaces and coupled redox processes that could contribute about one volt to the overall system,3 beside the pH and temperature gradients which contribute ∼300 millivolts.8c,20 Similarly, chemi-osmotic gradients are required for both modern acetogenesis and methanogenesis, suggesting that ancient processes may have used chemi-osmotic coupling mechanisms naturally existing at alkaline vents.21 Moreover, as shown by others, the products obtained in our study can be transformed into key biomolecules through reaction with ammonia and phosphates, reactants which are reported to be present around such vent systems.21,22 Thus, our combined experimental and computational study provides evidence that the crucial first step in this pathway from CO2 to biomolecules is feasible, and it therefore strongly supports the hypothesis that alkaline hydrothermal vent systems provided one possible environment for the pre-biotic chemistry preceding the onset of life.
The authors acknowledge the UK Engineering and Physical Sciences Research Council (EPSRC), grant numbers EP/H046313 and EP/K035355, for funding. Roldan thanks the Ramsay Memorial Fellowship Trust for a research fellowship and Islam acknowledges the UCL Industrial Doctorate Centre for Molecular Modelling and Materials Science, EPSRC (grant number EP/G036675) and the Dutch-Belgian Beamline at the ESRF (DUBBLE) for funding of a studentship. De Leeuw thanks the Royal Society of an Industry Fellowship. We have made use of HECToR and ARCHER via our membership of the UK's HPC Materials Chemistry Consortium (grants EP/L000202 and EP/F067496) as well as the UCL Legion High Performance Computing (Legion@UCL). We thank Dr Nikolaos Dimitratos for useful discussions.
Notes and references
- (a) P. Baaske, F. M. Weinert, S. Duhr, K. H. Lemke, M. J. Russell and D. Braun, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9346 CrossRef CAS PubMed; (b) E. V. Koonin and W. Martin, Trends Genet., 2005, 21, 647 CrossRef CAS PubMed; (c) J. H. E. Cartwright, J. M. Garcia-Ruiz, M. L. Novella and F. Otalora, J. Colloid Interface Sci., 2002, 256, 351 CrossRef CAS; (d) W. Martin and M. J. Russell, Philos. Trans. R. Soc. London, Ser. B, 2003, 358, 59 CrossRef CAS PubMed.
- M. J. Russell, R. M. Daniel, A. J. Hall and J. A. Sherringham, J. Mol. Evol., 1994, 39, 231 CrossRef CAS.
- M. J. Russell and A. J. Hall, J. Geol. Soc., 1997, 154, 377 CrossRef CAS.
- D. S. Kelley, J. A. Baross and J. R. Delaney, Annu. Rev. Earth Planet. Sci., 2002, 30, 385 CrossRef CAS.
- (a) J. B. Corliss, J. Dymond, L. I. Gordon, J. M. Edmond, R. P. V. Herzen, R. D. Ballard, K. Green, D. Williams, A. Bainbridge, K. Crane and T. H. Vanandel, Science, 1979, 203, 1073 CAS; (b) S. Humphris and U. American Geophysical, Seafloor hydrothermal systems: physical, chemical, biological, and geological interactions, American Geophysical Union, 1995 Search PubMed.
- (a) D. S. Kelley, J. A. Karson, G. L. Fruh-Green, D. R. Yoerger, T. M. Shank, D. A. Butterfield, J. M. Hayes, M. O. Schrenk, E. J. Olson, G. Proskurowski, M. Jakuba, A. Bradley, B. Larson, K. Ludwig, D. Glickson, K. Buckman, A. S. Bradley, W. J. Brazelton, K. Roe, M. J. Elend, A. Delacour, S. M. Bernasconi, M. D. Lilley, J. A. Baross, R. T. Summons and S. P. Sylva, Science, 2005, 307, 1428 CrossRef CAS PubMed; (b) G. L. Fruh-Green, D. S. Kelley, S. M. Bernasconi, J. A. Karson, K. A. Ludwig, D. A. Butterfield, C. Boschi and G. Proskurowski, Science, 2003, 301, 495 CrossRef PubMed; (c) G. Proskurowski, M. D. Lilley, D. S. Kelley and E. J. Olson, Chem. Geol., 2006, 229, 331 CrossRef CAS PubMed.
- G. Wachtershauser, Prog. Biophys. Mol. Biol., 1992, 58, 85 CrossRef CAS.
- (a) M. J. Russell and W. Martin, Trends Biochem. Sci., 2004, 29, 358 CrossRef CAS PubMed; (b) G. Wachtershauser, Chem. Biodiversity, 2007, 4, 584 CrossRef PubMed; (c) N. Lane and W. Martin, Cell, 2012, 151, 1406 CrossRef CAS PubMed.
- U. P. Apfel and W. Weigand, Angew. Chem., Int. Ed., 2011, 50, 4262 CrossRef CAS PubMed.
- H. Seino and M. Hidai, Chem. Sci., 2011, 2, 847 RSC.
- (a) W. Nitschke, S. E. McGlynn, E. J. Milner-White and M. J. Russell, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 871 CrossRef CAS PubMed; (b) H. Dobbek, V. Svetlitchnyi, L. Gremer, R. Huber and O. Meyer, Science, 2001, 293, 1281 CrossRef CAS PubMed; (c) I. Tsujisho, M. Toyoda and Y. Amao, Catal. Commun., 2006, 7, 173 CrossRef CAS PubMed; (d) T. Reda, C. M. Plugge, N. J. Abram and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654 CrossRef CAS PubMed; (e) A. Alissandratos, H. K. Kim, H. Matthews, J. E. Hennessy, A. Philbrook and C. J. Easton, Appl. Environ. Microbiol., 2013, 79, 741 CrossRef CAS PubMed; (f) H. Dobbek, V. Svetlitchnyi, J. Liss and O. Meyer, J. Am. Chem. Soc., 2004, 126, 5382 CrossRef CAS PubMed.
- A. Yamaguchi, M. Yamamoto, K. Takai, T. Ishii, K. Hashimoto and R. Nakamura, Electrochim. Acta, 2014, 141, 311 CrossRef CAS PubMed.
- M. G. Vladimirov, Y. F. Ryzhkov, V. A. Alekseev, V. A. Bogdanovskaya, V. A. Otroshchenko and M. S. Kritsky, Origins Life Evol. Biospheres, 2004, 34, 347 CrossRef CAS.
- W. Heinen and A. M. Lauwers, Origins Life Evol. Biospheres, 1996, 26, 131 CrossRef CAS.
- (a) N. Hollingsworth, A. Roffey, H.-U. Islam, M. Mercy, A. Roldan, W. Bras, M. Wolthers, C. R. A. Catlow, G. Sankar, G. Hogarth and N. H. de Leeuw, Chem. Mater., 2014, 26, 6281 CrossRef CAS; (b) H.-U. Islam, A. Roffey, N. Hollingsworth, R. Catlow, M. Wolthers, N. De Leeuw, W. Bras, G. Sankar and G. Hogarth, 15th International Conference on X-Ray Absorption Fine Structure (Xafs15), 2013, 430; (c) J. C. Bear, N. Hollingsworth, P. D. McNaughter, A. G. Mayes, M. B. Ward, T. Nann, G. Hogarth and I. P. Parkin, Angew. Chem., Int. Ed., 2014, 53, 1598 CrossRef CAS PubMed.
- D. Santos-Carballal, A. Roldan, R. Grau-Crespo and N. H. de Leeuw, Phys. Chem. Chem. Phys., 2014, 16, 21082 RSC.
- (a) H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225 CrossRef; (b) J. P. Pradiser and C. M. Pradier, Carbon Dioxide Chemistry: Environmental Issues, Woodhead Publishing, 1994 Search PubMed.
- (a) T. M. McCollom and J. S. Seewald, Chem. Rev., 2007, 107, 382 CrossRef CAS PubMed; (b) T. M. McCollom and J. S. Seewald, Geochim. Cosmochim. Acta, 2003, 67, 3625 CrossRef CAS.
- W. Martin, J. Baross, D. Kelley and M. J. Russell, Nat. Rev. Microbiol., 2008, 6, 805 CAS.
- G. Macleod, C. McKeown, A. Hall and M. Russell, Origins Life Evol. Biospheres, 1994, 24, 19 CrossRef CAS.
- W. Martin and M. J. Russell, Philos. Trans. R. Soc., B, 2007, 362, 1887 CrossRef CAS PubMed.
- (a) R. Egel, D.-H. Lankenau and A. Y. Mulkidjanian, Origins of Life: The Primal Self-Organization, Springer, Berlin, Heidelberg, 2011 Search PubMed; (b) R. Saladino, C. Crestini, S. Pino, G. Costanzo and E. Di Mauro, Phys. Life Rev., 2012, 9, 84 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, 1H NMR spectra and other characterization data mentioned in the text. See DOI: 10.1039/c5cc02078f |
| This journal is © The Royal Society of Chemistry 2015 |