
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of chiral cyclobutanes via rhodium/diene-catalyzed asymmetric 1,4-addition: a dramatic ligand effect on the diastereoselectivity†
Ya-Jing
Chen
,
Tian-Jiao
Hu
,
Chen-Guo
Feng
* and
Guo-Qiang
Lin
CAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P. R. China. E-mail: fengcg@sioc.ac.cn; Fax: +86 21 5080 7388; Tel: +86 21 5492 5081
First published on 17th April 2015
Abstract
A highly diastereo- and enantioselective rhodium-catalyzed arylation of cyclobutenes for the efficient synthesis of chiral cyclobutanes has been developed. Chiral diene ligands exhibited excellent capability for the reaction diastereoselectivity control.
Chiral cyclobutanes exist as core subunits in numerous natural products1,2 and pharmaceutical agents.3 Moreover, a number of interesting chemical transformations associated with the four-membered strained ring render them valuable building blocks for organic synthesis.4 As a result, significant efforts have been directed to the asymmetric construction of chiral cyclobutane rings,5 and some catalytic asymmetric methods have been developed during the past decades, including asymmetric organo-6 or Lewis acid-catalyzed [2+2] cycloaddition,7 asymmetric ring expansion of cyclopropanes,8 asymmetric intramolecular ring closure of linear substrates9 and asymmetric functionalization of prochiral cyclobutane derivatives.10–12 The last strategy can avoid the relatively difficult construction of the constrained ring. More importantly, it provides a diverse synthetic approach using different functionalization reactions and easily accessible cyclobutanes as starting materials. To the best of our knowledge, almost all the present successful examples are limited to the chemical modifications of cyclobutanone substrates (Scheme 1, eqn (1)).10 Very recently, Yu and co-workers reported the asymmetric C–H activation/arylation of cyclobutane rings (eqn (2)).11 Maulide and co-workers reported the palladium-catalyzed allylic alkylation of 4-chlorocyclobut-2-enecarboxylic acid (eqn (3)).12 We envisioned that the well-established asymmetric rhodium-catalyzed addition of organometallic reagents to electron deficient double bonds could lead to a straightforward and diverse access to chiral cyclobutanes.13 Although five- and six-membered counterparts are among the most tested substrates in rhodium-catalyzed 1,4-additions, only a single non-asymmetric example of the addition to cyclobutene was introduced by Tang and co-workers14 in their synthesis toward pipercyclobutanamide A and piperchabamide G and its asymmetric version remains unreported.
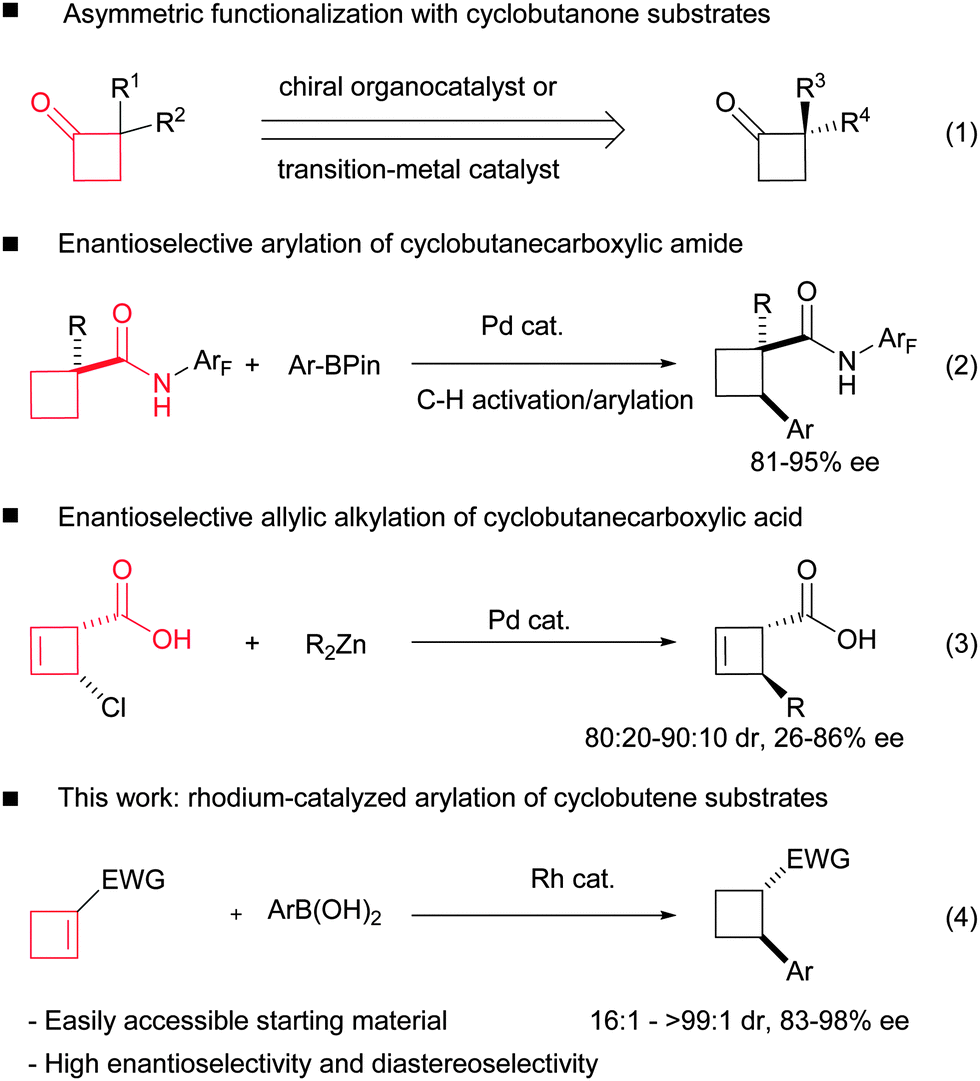 | ||
| Scheme 1 Asymmetric functionalization of cyclobutane derivatives. | ||
We reason that cyclobutene-1-carboxylate esters are suitable precursors for the enantioselective synthesis of chiral cyclobutanes, which can be easily prepared via three steps from widely available ethyl cyclobutanecarboxylate,15 and the ester groups could serve as a handle for further functionalization. However, it would be challenging to control both stereogenic centers generated in the addition process with such substrates. Especially, upon the first enantioselective addition at the β-position, the subsequent protonation of the resulting oxa-π-allylrhodium intermediate would generate a second chiral center, which is usually quite difficult to control unless the α-substituent can induce a different protonation pathway in the reported similar examples.16 Herein, we report our research on the ligand enabled highly diastereo- and enantioselective rhodium-catalyzed arylation of cyclobutene-1-carboxylate esters for the efficient synthesis of chiral cyclobutanes.
First, we tested the addition of phenylboronic acid to the ethyl ester substrate (1a) in the presence of the active hydroxo rhodium complex [Rh(OH)(L1)].17 Gratifyingly, the reaction took place smoothly, affording the desired product in 61% yield with high diastereoselectivity despite low enantioselectivity. It was reported that the ester substituents could affect the stereoselectivity control for the aryl addition to linear α,β-unsaturated ester systems,18 thus a variety of different esters were examined in our case (Table 1). Although more sterically hindered substituents clearly improved the enantioselectivity, the reaction yield and diastereoselectivity were highly dependent on the structure pattern of substituents. The 2,6-diphenylphenyl substituent provided the highest enantioselectivity and maintained high diastereoselectivity, but a reduced reaction yield (entry 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate (R) | Yieldb (%) | trans/cisc | trans-3 | ee of 3d (%) |
| a Reactions were carried out with 1 (0.1 mmol), 2a (0.2 mmol), [Rh(OH)(L1)]2 (0.0015 mmol), 2.0 M aq. KOH (0.2 mmol) in toluene (1 mL) at 50 °C for 5 h. b A combined isolated yield of 3 and 4. c Determined by GC analysis. d Determined by chiral HPLC analysis. | |||||
| 1 | 1a (Et) | 61 | 13/1 | 3aa | 21 |
| 2 | 1b (iPr) | 50 | 12/1 | 3ba | 24 |
| 3 | 1c (tBu) | 50 | 5/1 | 3ca | 74 |
| 4 | 1d (Bn) | 21 | 12/1 | 3da | 9 |
| 5 | 1e (Ph) | 25 | 15/1 | 3ea | 74 |
| 6 | 1f (2,6-Me2C6H3) | 80 | 2.6/1 | 3fa | 87 |
| 7 | 1g (2,6-iPr2C6H3) | 99 | 2.1/1 | 3ga | 89 |
| 8 | 1h (2,6-Ph2C6H3) | 35 | 12/1 | 3ha | 92 |
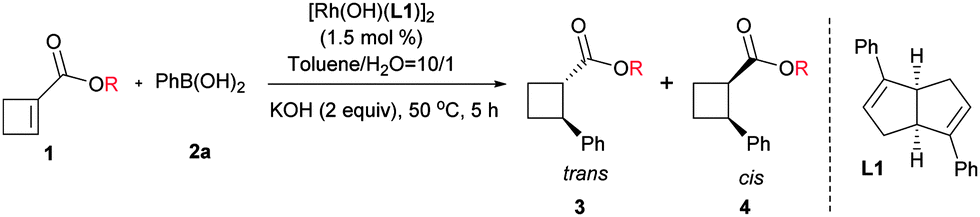
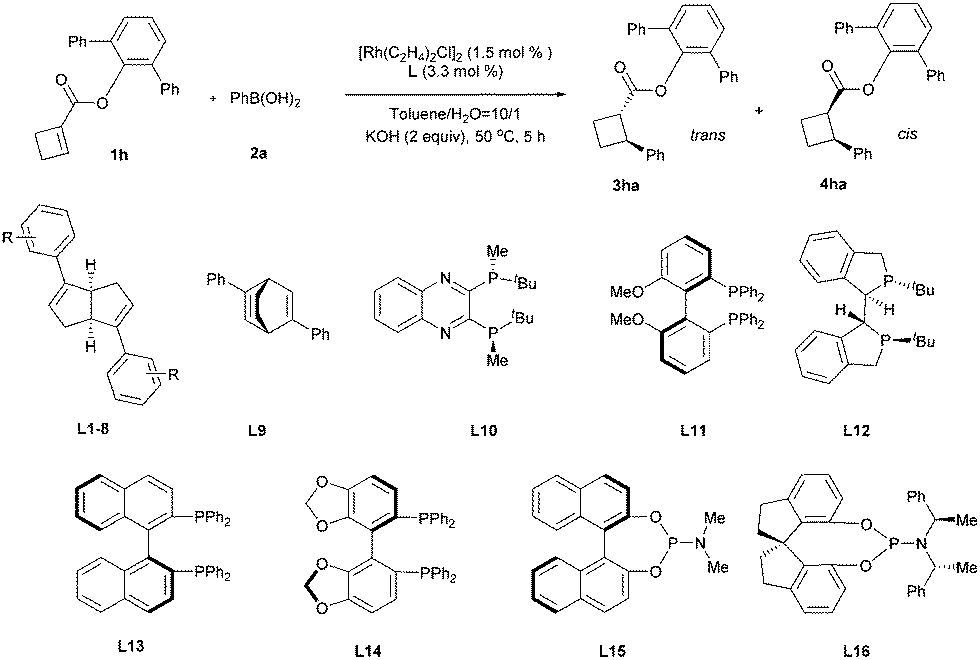
Next, we turned to investigate this reaction using an in situ generated chiral rhodium catalyst. With ester 1h as a substrate and [RhCl(C2H4)2]2 as a precatalyst, a variety of chiral ligands were examined (Table 2).19 A significantly improved yield was obtained from the same ligand L1 (entry 1 vs.Table 1, entry 8). However, attempts to further increase the enantioselectivity of this reaction by introducing different substituents to the phenyl ring of ligand L1 were unsuccessful. It is worth mentioning that the diastereoselectivity was significantly improved by the introduction of electron-withdrawing substituents (entries 2–9). The highest diastereoselectivity was achieved using para 4-CF3O substituted ligand L8, albeit with a slight loss in reaction yield, which was brought back by a lower temperature and a prolonged reaction time (entries 8 and 9). Bicyclo[2.2.2]octadiene L920 gave a similar reaction yield and diastereoselectivity compared with L1, but with lower enantioselectivity (entry 10). All the tested phosphine-containing ligands failed to promote this reaction effectively and only a trace amount of the product was generated in most cases under current reaction conditions (entries 11–17). Interestingly, while bisphosphine ligands gave low diastereoselectivities, relatively electron-deficient phosphoramidite ligands showed much higher diastereoselectivities, consistent with the trend observed for diene ligands. The high diastereoselectivity control by electron-deficient ligands may be attributed to the quick protonation of the carbon–rhodium bond, which was generated after the cis addition of the aryl rhodium species to the olefin. The stereochemically retentive direct protonation step could prevent the uncontrollable oxa-π-allylrhodium protonation pathway,21 thus the addition reaction led to the trans product.
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Yieldb (%) | trans/cisc | ee of 3had (%) |
| a Reactions were carried out with 1h (0.1 mmol), 2a (0.2 mmol), [Rh(C2H4)2Cl]2 (0.0015 mmol), L (0.0033 mmol), 2.0 M aq. KOH (0.2 mmol) in toluene (1 mL) at 50 °C for 5 h unless otherwise noted. b A combined yield of 3ha and 4ha, which was determined by GC using dodecane as an internal standard. c Determined by GC. d Determined by chiral HPLC analysis. e Reaction was made to run at 30 °C for 24 h. | ||||
| 1 | L1 (R = H) | 97 | 15/1 | 92 |
| 2 | L2 (R = 4-MeO) | 77 | 11/1 | 89 |
| 3 | L3 (R = 3,5-Me2) | 73 | 9/1 | 93 |
| 4 | L4 (R = 3,5-MeO2) | 74 | 10/1 | 91 |
| 5 | L5 (R = 4-F) | 88 | 20/1 | 88 |
| 6 | L6 (R = 4-CF3) | 80 | 40/1 | 92 |
| 7 | L7 (R = 3,5-F2) | 75 | 47/1 | 92 |
| 8 | L8 (R = 4-OCF3) | 72 | 52/1 | 92 |
| 9e | L8 (R = 4-OCF3) | 99 | 62/1 | 92 |
| 10 | L9 | 86 | 14/1 | 74 |
| 11 | L10 | 8 | 2.2/1 | — |
| 12 | L11 | 4 | 1.5/1 | — |
| 13 | L12 | 1 | 1.6/1 | — |
| 14 | L13 | 21 | 1.3/1 | 96 |
| 15 | L14 | 5 | 1.5/1 | — |
| 16 | L15 | 18 | 8/1 | 25 |
| 17 | L16 | 3 | 25/1 | — |
With optimal reaction conditions identified (Table 2, entry 9), a wide range of arylboronic acids were explored as depicted in Table 3. Excellent diastereoselectivities were obtained for all the tested arylboronic acids although the reaction yields were affected by both steric and electronic properties of aryl groups. In general, electron-donating groups at the para position of the phenyl ring would benefit the reaction yields, and the sterically hindered ortho-substituted arylboronic acid gave only moderate reaction yield even at elevated reaction temperature. As for the enantioselectivity, the reactions with most arylboronic acids provided good to excellent ees (83–98%). The absolute configuration of product 3hh was ambiguously determined by a single-crystal X-ray analysis as depicted in Scheme 2,22 and other products were assigned by analogy.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | trans-3 | Yieldb (%) | trans/cisc | eed (%) |
| a Reactions were carried out with 1h (0.2 mmol), 2 (0.4 mmol), [Rh(C2H4)2Cl]2 (0.003 mmol), L8 (0.0066 mmol), 2.0 M aq. KOH (0.4 mmol) in toluene (2 mL) at 30 °C for 24 h unless otherwise noted. b Isolated yield. c Determined by GC. d Determined by chiral HPLC analysis. e 50 °C. | |||||
| 1 | Ph (2a) | 3ha | 99 | 62/1 | 92 |
| 2 | 4-MeOC6H4 (2b) | 3hb | 90 | 99/1 | 89 |
| 3 | 4-tBuC6H4 (2c) | 3hc | 97 | 49/1 | 93 |
| 4 | 4-CF3C6H4 (2d) | 3hd | 73 | 32/1 | 98 |
| 5e | 4-CO2MeC6H4 (2e) | 3he | 36 | >99/1 | 92 |
| 6 | 4-FC6H4 (2f) | 3hf | 78 | 32/1 | 97 |
| 7 | 4-ClC6H4 (2g) | 3hg | 96 | 49/1 | 97 |
| 8 | 4-BrC6H4 (2h) | 3hh | 92 | 49/1 | 96 |
| 9 | 3-MeOC6H4 (2i) | 3hi | 91 | >99/1 | 93 |
| 10 | 3-FC6H4 (2j) | 3hj | 84 | 49/1 | 97 |
| 11 | 3-CO2MeC6H4 (2k) | 3hk | 95 | 32/1 | 96 |
| 12e | 2-MeO (2l) | 3hl | 51 | 16/1 | 96 |
| 13 | 1-Naphthyl (2m) | 3hm | 96 | >99/1 | 83 |
| 14 | 2-Naphthyl (2n) | 3hn | 68 | >99/1 | 87 |
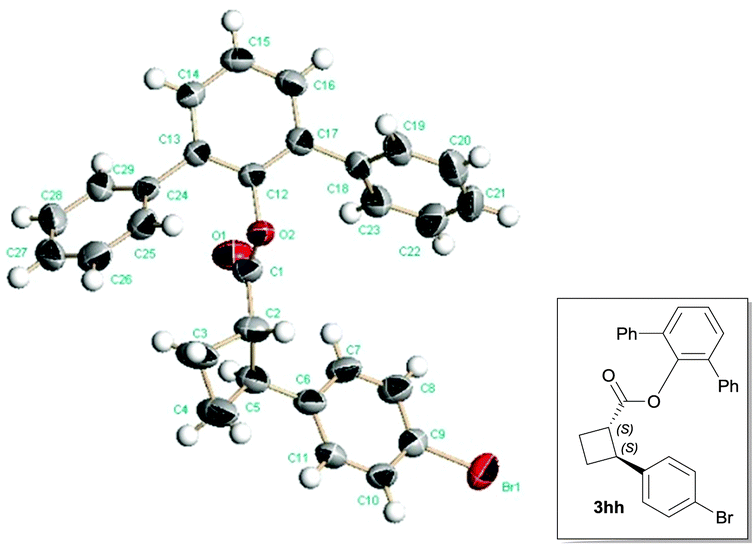 | ||
| Scheme 2 The X-ray diffraction structure of 3hh. | ||
The obtained products can be easily converted to other cyclobutane derivatives. As shown in Scheme 3, reduction of ester 3ha with LiAlH4 at room temperature furnished alcohol 5 in 97% yield. The hydrolysis could provide acid 6 in 89% yield. Treatment of ester 3ha with lithium diisopropylamide (LDA) followed by alkylation with allyl iodide afforded product 7 as a single diastereomer with a negligible loss in optical purity.
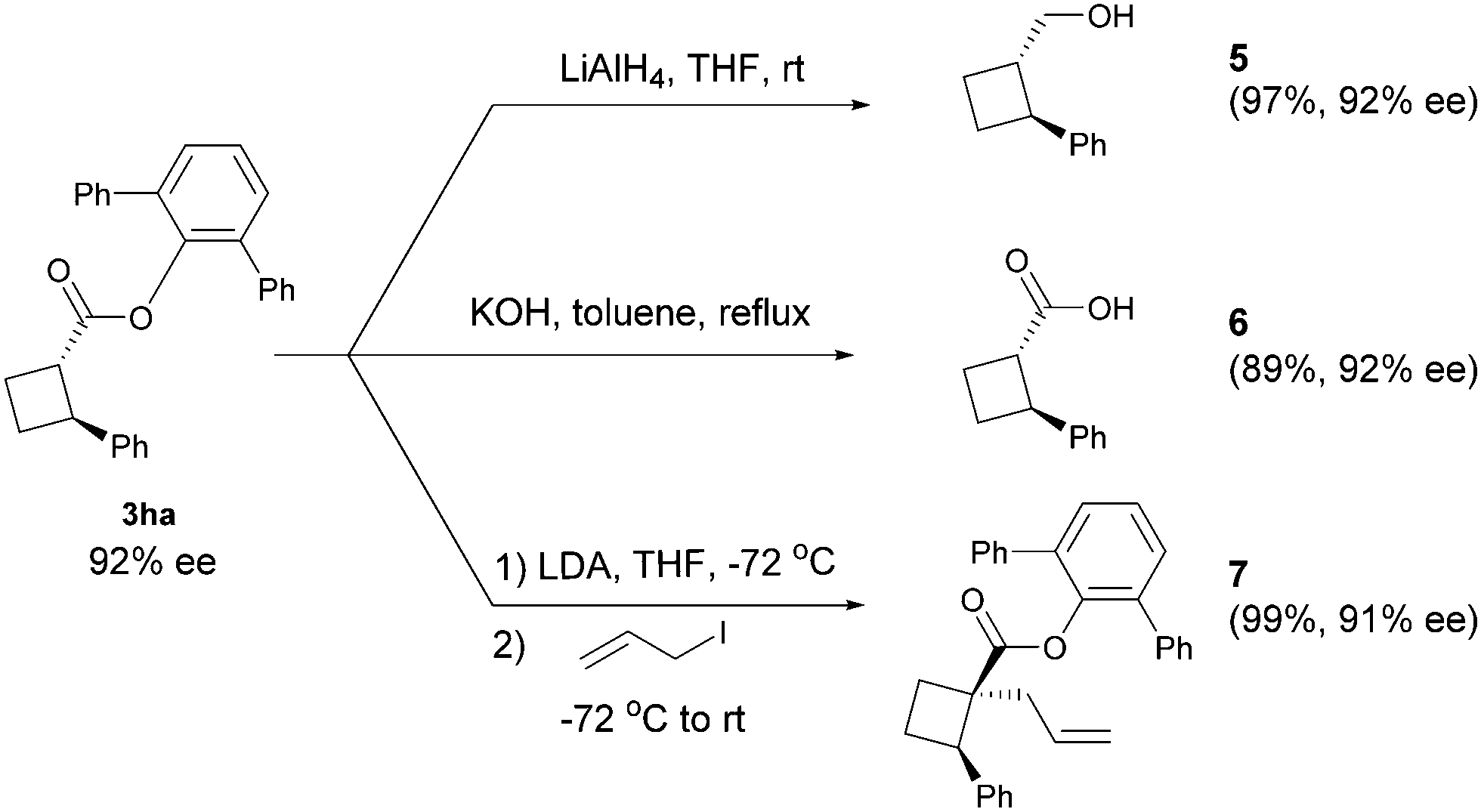 | ||
| Scheme 3 Transformations of product 3ha. | ||
In summary, we have developed a highly diastereo- and enantioselective rhodium-catalyzed arylation of cyclobutene-1-carboxylate esters, which provides an efficient synthetic approach for chiral butanes. Chiral diene ligands, especially electron deficient ones, exhibited excellent diastereoselectivity control. In addition, the versatile transformations of the product warrant the synthetic utility of this methodology.
This work was generously supported by the National Natural Science Foundation of China (21472212), the Shanghai Municipal Committee of Science and Technology (14ZR1448500) and the Collaborative Innovation Center of Chemical Science and Engineering (Tianjing). We acknowledge Dr Han-Qing Dong for his help in the preparation of this manuscript.
Notes and references
- For review: V. M. Dembitsky, J. Nat. Med., 2008, 62, 1–33 CrossRef CAS PubMed
.
- For selected examples:
(a) P. S. Baran, D. P. O'Malley and A. L. Zografos, Angew. Chem., Int. Ed., 2004, 43, 2674–2677 CrossRef CAS PubMed
- For selected examples:
(a) J. Singh, G. S. Bisacchi, S. Ahmad, J. D. Godfrey, Jr., T. P. Kissick, T. Mitt, O. Kocy, T. Vu, C. G. Papaioannou, M. K. Wong, J. E. Heikes, R. Zahler and R. H. Mueller, Org. Process Res. Dev., 1998, 2, 393–399 CrossRef CAS
- For review:
(a) J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485–1537 CrossRef CAS PubMed
- For review: E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449–1483 CrossRef CAS PubMed
-
(a) K. Ishihara and K. Nakano, J. Am. Chem. Soc., 2007, 129, 8930–8931 CrossRef CAS PubMed
-
(a) T. A. Engler, M. A. Letavic, R. Iyengar, K. O. LaTessa and J. P. Reddy, J. Org. Chem., 1999, 64, 2391–2405 CrossRef CAS
- F. Kleinbeck and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 9178–9179 CrossRef CAS PubMed
-
(a) R. Panish, S. R. Chintala, D. T. Boruta, Y. Z. Fang, M. T. Taylor and J. M. Fox, J. Am. Chem. Soc., 2013, 135, 9283–9286 CrossRef CAS PubMed
-
(a) M. Lopp, A. Paju, T. Kanger and T. Pehk, Tetrahedron Lett., 1996, 37, 7583–7586 CrossRef CAS
- K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138–8142 CrossRef CAS PubMed
- A. Misale, S. Niyomchon, M. Luparia and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 7068–7073 CrossRef CAS PubMed
- For leading reviews on Rh-catalyzed asymmetric conjugated additions:
(a) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed
- R. Liu, M. Zhang, T. P. Wyche, G. N. Winston-McPherson, T. S. Bugni and W. Tang, Angew. Chem., Int. Ed., 2012, 51, 7503–7506 CrossRef CAS PubMed
-
(a) A. Campbell and H. N. Rydon, J. Chem. Soc., 1953, 3002–3008 RSC
-
(a) T. Hayashi, T. Senda and M. Ogasawara, J. Am. Chem. Soc., 2000, 122, 10716–10717 CrossRef CAS
-
(a) Z.-Q. Wang, C.-G. Feng, M.-H. Xu and G.-Q. Lin, J. Am. Chem. Soc., 2007, 129, 5336–5337 CrossRef CAS PubMed
-
(a) J.-F. Paquin, C. R. J. Stephenson, C. Defieber and E. M. Carreira, Org. Lett., 2005, 7, 3821–3824 CrossRef CAS PubMed
- Reviews of chiral olefin ligands:
(a) C. Defieber, H. Grüetzmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482–4502 CrossRef CAS PubMed
- N. Tokunaga, Y. Otomaru, K. Okamoto, K. Ueyama, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2004, 126, 13584–13585 CrossRef CAS PubMed
- C. M. So, S. Kume and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 10990–10993 CrossRef CAS PubMed
- CCDC 1042990 contains the supplementary crystallographic data for compound 3hh.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1042990. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc02023a |
| This journal is © The Royal Society of Chemistry 2015 |