
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanochemical reactions studied by in situ Raman spectroscopy: base catalysis in liquid-assisted grinding†
Martina
Tireli‡
a,
Marina
Juribašić Kulcsár‡
a,
Nikola
Cindro
b,
Davor
Gracin
a,
Nikola
Biliškov
a,
Mladen
Borovina
ab,
Manda
Ćurić
a,
Ivan
Halasz
*a and
Krunoslav
Užarević
*a
aRuđer Bošković Institute, Bijenička 54, HR-10000 Zagreb, Croatia. E-mail: ivan.halasz@irb.hr; krunoslav.uzarevic@irb.hr
bDepartment of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, HR-10000 Zagreb, Croatia
First published on 1st April 2015
Abstract
In situ Raman spectroscopy was employed to study the course of a mechanochemical nucleophilic substitution on a carbonyl group. We describe evidence of base catalysis, akin to catalysis in solution, achieved by liquid-assisted grinding.
Organic synthesis is traditionally devoted to solution techniques for performing chemical reactions. However, solid-state reactions achieved by milling have emerged as an alternative to solution reactions.1–3 Various organic reactions have been reported to occur under mechanochemical or solvent-free reaction conditions such as Suzuki4a and Sonogashira4b reactions, Michael addition,5a Diels–Alder reaction,5b including solid-state functionalization of graphene nanoplatelets,5c and Wittig reactions,5d as well as multi-step and one-pot reactions.6 The scope of mechanochemical reactivity is significantly broadened by techniques such as liquid-assisted grinding (LAG)7 where additives in sub-stoichiometric amounts have been found to improve the reactivity.8 While the mechanism of their action is generally unknown, additives were found to act as catalysts9 and may steer the reaction towards a specific product.10 Also, it was recently indicated that a one-pot mechanochemical Gewald reaction can be catalysed by base.11
Contrary to well understood mechanisms of organic reactions in solution, mechanistic understanding of mechanochemical organic reactions has thus far remained elusive. This limits the use and applicability of these solid-state processes and their possible development for industrial purposes. Thus far, mechanisms of organic mechanochemical reactions were mainly deduced from stepwise ex situ analysis which provides only limited information due to an inherent time delay between sampling and analysis but also due to the fact that sampling may irreproducibly disturb the mechanochemical process. Recently however, two techniques based on powder X-ray diffraction (PXRD)12 or Raman spectroscopy13 were introduced to study mechanochemical reactions in situ and in real time without the need to interrupt the milling process. The two techniques are complementary and are ideally employed simultaneously.14 While PXRD is suitable for monitoring the evolution of bulk crystalline phases, Raman spectroscopy may be more appropriate for monitoring of amorphous or liquid phases which are particularly common in organic solid-state reactions.15
Here, we have employed in situ Raman spectroscopy to study the effect of additives on a mechanochemical organic reaction. Using the reaction between 4-nitrobenzoyl azide (1) and 1,4-diaminobenzene (2) yielding N-(4-aminophenyl)-4-nitrobenzamide (3) (Scheme 1) as a model nucleophilic substitution reaction on the carbonyl group, we have examined various additives differing in proticity, polarity and acid/base properties. Performed experiments aimed to establish whether the effect of additives is merely physical, through dissolution or enhanced molecular mobility,16 or if the additives actively participate through chemical interactions.
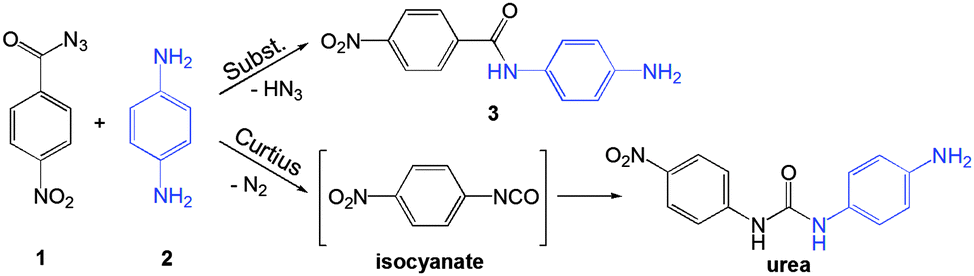 | ||
| Scheme 1 Nucleophilic substitution on the carbonyl group of an acyl azide and potential Curtius rearrangement yielding an isocyanate intermediate which reacts with the amine to yield the urea derivative. | ||
Acyl azides are usually exploited in Curtius rearrangement where the acyl azide group rearranges to the isocyanate group upon exposure to mild external stimuli such as heat or irradiation. Curtius rearrangement during milling could be expected, based on the latest studies of energetics of mechanochemical treatment,17 but the urea product that would result from the reaction of the amine with the isocyanate intermediate was not observed.18 Instead, the azide here reacted with the amine to quantitatively yield the corresponding amide (Scheme 1). However, focus of this work is not mechanochemical formation of the amide group, for which other synthetic procedures under mechanochemical treatment were recently described,19 but rather the mechanistic aspect of nucleophilic attack on the carbonyl group.
Time-resolved Raman spectra recorded in situ during milling (Fig. 1) were evaluated using two-dimensional (2D) correlation analysis according to the procedure described by Noda and Ozaki.20 Both the synchronous and asynchronous spectra of all investigated systems exhibited stark similarities (Fig. S6–S9 in ESI†). Namely, both the diagonal and cross bands behave in the same way, which allows direct comparison of reactions under different reaction conditions. Assignment of the most relevant spectral features was done according to calculated Raman spectra21 (Fig. 2) and previously published spectra of 222 and compounds similar to 123 (Table S1 in the ESI†). Formation of 3 in Fig. 1 is characterised by changes in the region between ca. 1600 and 1700 cm−1 where it has three partially overlapping bands (Fig. 2). The amide II band at 1525 cm−1 and the amide III band at 1268 cm−1 emerge while the azide carbonyl stretching band at 1694 cm−1 diminishes during milling. The band belonging to symmetric nitro group vibration at 1352 cm−1 remains unaltered during the reaction. Curtius rearrangement would here result in 4-nitrophenyl isocyanate as an intermediate which would subsequently yield a derivative of urea (Scheme 1). The characteristic ν(CN) band of isocyanate expected at 2320 cm−1 was, however, not observed (Fig. S10 in the ESI†).
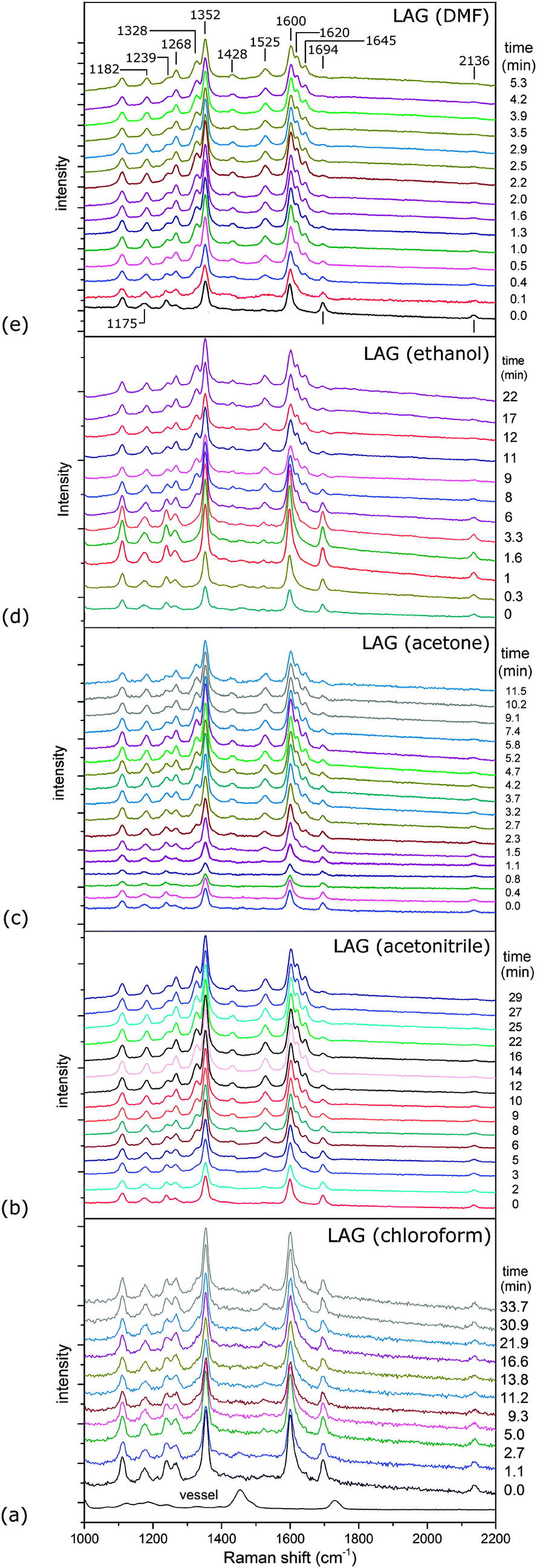 | ||
| Fig. 1 Time-resolved Raman spectra recorded under different LAG conditions using as liquid additives: (a) chloroform, (b) acetonitrile, (c) acetone, (d) ethanol and (e) N,N-dimethylformamide (DMF). The spectrum of the reaction vessel is given in (a). Band positions are depicted in (e). | ||
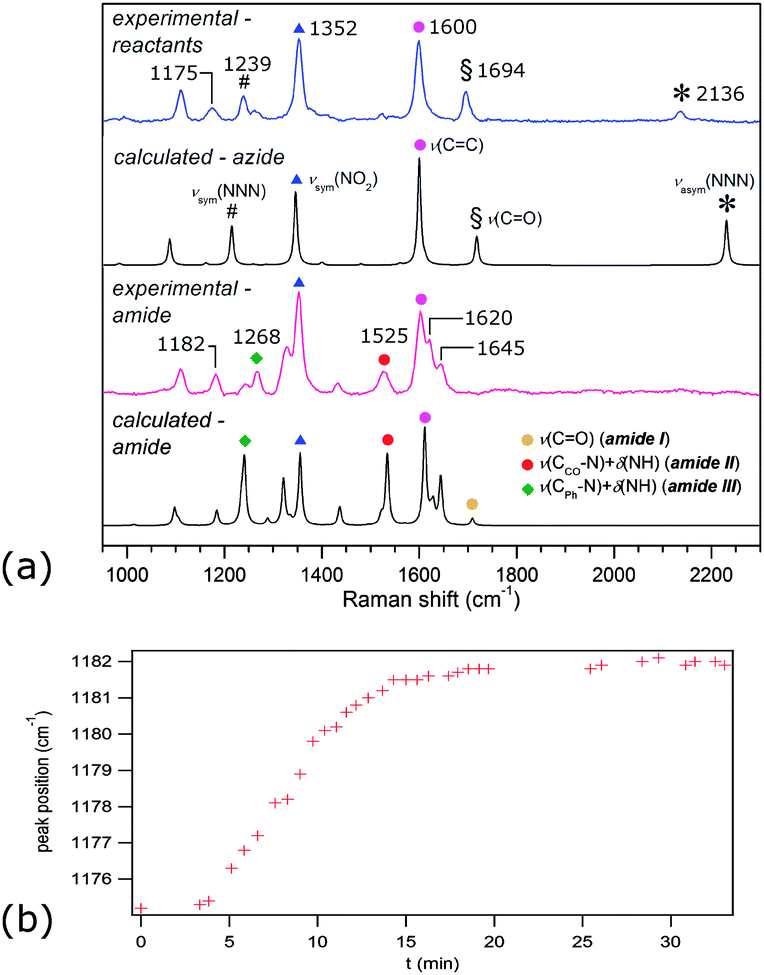 | ||
| Fig. 2 (a) Observed and calculated Raman spectra of participating species with band positions and assignments. Raman scattering of the amine was very weak in comparison to scattering of azide and amide. (b) Band shift during LAG reaction with acetonitrile. | ||
The intensity of the band at 1239 cm−1, assigned to symmetric stretching of the azide group, initially decreases during milling but does not diminish completely (Fig. S11 in the ESI†). Simultaneously, the band at 1175 cm−1, which changes its position during milling to 1182 cm−1 (Fig. 2b), and the band at 1266 cm−1 increase in intensity, reflecting the formation of 3. These bands are positively correlated in 2D synchronous spectra, but their correlation with the band at 1239 cm−1 is absent (Fig. S6–S9 in the ESI†). However, cross peaks in asynchronous spectra show that the change in the 1239 cm−1 band occurs before changes in bands at 1175 and 1266 cm−1. Interestingly, the intensity of the 2136 cm−1 band, which is the antisymmetric counterpart of the 1239 cm−1 band, continuously drops during the reaction. This indicates another emerging species contributing to the intensity of the band at 1239 cm−1. At first sight, no evidence could be found for the formation of hydrazoic acid (HN3) which is formally the leaving group in the substitution reaction (Scheme 1). This could have been explained by its volatility and possible degradation resulting from mechanical shock from milling ball impacts.24 However, we note that the band at 1239 cm−1, which survives the whole milling process, could tentatively belong to HN3, supporting the assumed reaction pathway toward amide without intermediate species, where HN3 is formed as a by-product.
Neat milling25 of 1 and 2 for 60 minutes resulted in slow and incomplete formation of 3 (Fig. S12 in the ESI†). Liquid-assisted grinding was, in comparison, far more successful in driving the amide formation (Fig. 1, Table 1). Except for chloroform which had little effect, liquid additives dramatically increased the reaction rate. This was especially true for LAG using DMF, followed by ethanol, acetone and acetonitrile. As stated above, all studied reactions proceeded along the same pathway, thus allowing examination of the effect of additives on the mechanochemical process. Since LAG with chloroform resulted in no or only minor reaction rate acceleration, while with other protic and polar liquid additives significantly accelerated the reaction, we conclude that the mere presence of the liquid to enhance the molecular mobility was not enough to facilitate product formation. This is also evident from the lack of a correlation between solubilities of participating species (Table S2 in the ESI†) in each solvent and the corresponding reaction times.
| Reaction time (min) | Basicity (DN)a | Dipole moment (D) | Relative permittivity | |
|---|---|---|---|---|
| a DN – donor number by Gutmann.26 b Reaction was not complete. c dabco is not a salt but a molecular compound. d We worked under the assumption that dabco and NaOAc increased the basicity while ammonium nitrate reduced the basicity relative to pure ethanol. | ||||
| LAG (liquid) | ||||
| DMF | 4 | 26.6 | 3.82 | 36.1 |
| Ethanol | 8 | 19.2 | 1.69 | 24.5 |
| Acetone | 10 | 17.0 | 2.88 | 20.7 |
| Acetonitrile | 16.5 | 14.1 | 3.92 | 38.0 |
| Chloroform | 30b | — | 1.15 | 4.81 |
| No liquid | 60b | — | — | — |
| ILAG (ethanol, salt) | ||||
| dabcoc | 6.5 | —d | — | — |
| NaOAc | 7.2 | —d | — | — |
| NH4NO3 | 8 | —d | — | — |
However, we note that product formation was faster when the liquid additive was more basic. This is clearly suggested from the correlation of the time needed for reaction completion and basicity of the liquid additive described using Gutmann's donor numbers26 (Table 1, Fig. 3). Such a correlation with reaction times could not be established using liquid's polarity, represented by the molecule dipole moment, or its relative permittivity (Table 1).
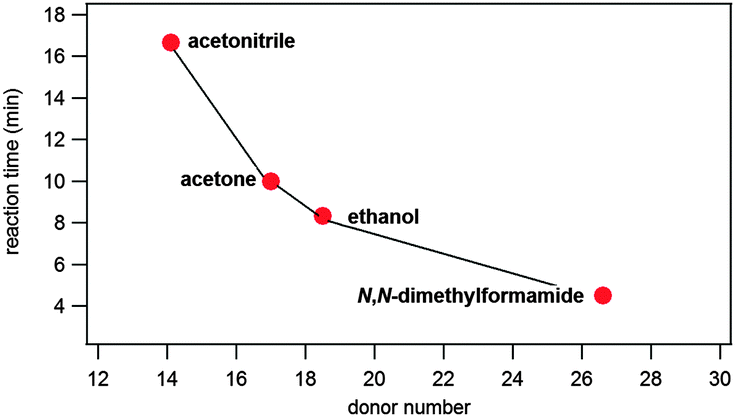 | ||
| Fig. 3 Correlation of reaction times and liquid basicity, expressed through Gutmann's donor numbers,26 in LAG reactions. | ||
To corroborate this, simple salts were introduced in the reaction mixture along with ethanol (Table S1 and Fig. S13–S15 in the ESI†). Resulting reaction conditions are termed ion- and liquid-assisted grinding (ILAG) and may offer further possibilities of reaction control.10b With basic sodium acetate (10 mol% with respect to reactants) added along with ethanol, the reaction was accelerated relative to pure LAG with ethanol. If an even stronger base like 1,4-diazabicyclo[2.2.2]octane (dabco) was used in an analogous experiment, the reaction was even more accelerated. If however, slightly acidic ammonium nitrate was added along with ethanol, the reaction rate remained essentially the same. This strongly supports the conclusion that this nucleophilic substitution reaction on a carbonyl group is base-catalysed and that proton abstraction is an important step in the reaction's mechanism.
In summary, we have employed in situ Raman spectroscopy for uninterrupted, real-time monitoring of a mechanochemical organic reaction. Nucleophilic substitution on the carbonyl group was conducted under various LAG conditions which revealed that the reaction is base-catalysed by the liquid additive. Such behaviour resembles acid/base catalysis as could be expected for solution reactions and indicates the possibility of reaction transfer to the liquid phase under LAG conditions.7c,27 The presented results not only demonstrate high sensitivity of organic mechanochemical reactions to additives, but also provide a new entry for systematic understanding and control of mechanochemical reactivity.
The authors thank the Ministry of Science, Educations and Sports of the Republic of Croatia for financial support (Grant No. 098-0982915-2950 and 098-0982886-2894). We are grateful to Mr Vitomir Stanišić and Mr Siniša Pecik for manufacturing PMMA reaction vessels and to Dr Aleksandar Sabljić for support. The LS2 laser source used in this study was kindly donated by PD-LD Inc., Pennington, New Jersey (USA). Computations were performed on the Isabella cluster at SRCE, Zagreb.
Notes and references
- (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC; (b) T. Friščić, Chem. Soc. Rev., 2012, 41, 3493 RSC.
- (a) G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668 RSC; (b) E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719 RSC; (c) G. Kaupp, CrystEngComm, 2009, 11, 388 RSC; (d) T. Friščić, I. Halasz, V. Štrukil, M. Eckert-Maksić and R. E. Dinnebier, Croat. Chem. Acta, 2012, 85, 367 CrossRef.
- (a) K. Komatsu, Top. Curr. Chem., 2005, 254, 185 CAS; (b) A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317 RSC; (c) A. Bruckmann, A. Krebs and C. Bolm, Green Chem., 2008, 10, 1131 RSC; (d) L. Konnert, F. Lamaty, J. Martinez and E. Colacino, J. Org. Chem., 2014, 79, 4008 CrossRef CAS PubMed.
- (a) F. Schneider and B. Ondruschka, ChemSusChem, 2008, 1, 622 CrossRef CAS PubMed; (b) R. Thorwirth, A. Stolle and B. Ondruschka, Green Chem., 2010, 12, 985 RSC.
- (a) Y.-F. Wang, R.-X. Chen, K. Wang, B.-B. Zhang, Z.-B. Li and D.-Q. Xu, Green Chem., 2012, 14, 893 RSC; (b) D. Huertas, M. Florscher and V. Dragojlovic, Green Chem., 2009, 11, 91 RSC; (c) J.-M. Seo, I.-Y. Jeona and J.-B. Baek, Chem. Sci., 2013, 4, 4273 RSC; (d) V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, J. Am. Chem. Soc., 2002, 124, 6244 CrossRef CAS PubMed.
- (a) B. Içli, N. Christinat, J. Tönneman, C. Schüttler, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2009, 131, 3154 CrossRef PubMed; (b) M. Ferguson, N. Giri, X. Huang, D. Apperley and S. L. James, Green Chem., 2014, 16, 1374 RSC; (c) D. Cinčić and T. Friščić, CrystEngComm, 2014, 16, 10169 RSC.
- (a) N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372 RSC; (b) T. Friščić, A. V. Trask, W. Jones and W. D. S. Motherwell, Angew. Chem., Int. Ed., 2006, 45, 7546 CrossRef PubMed; (c) E. A. Losev and E. V. Boldyreva, CrystEngComm, 2014, 16, 3857 RSC.
- (a) T. Friščić, I. Halasz, F. C. Strobridge, R. E. Dinnebier, R. S. Stein, L. Fábián and C. Curfs, CrystEngComm, 2011, 13, 3125 RSC; (b) F. C. Strobridge, N. Judaš and T. Friščić, CrystEngComm, 2010, 12, 2409 RSC.
- (a) J.-L. Do, C. Mottillo, D. Tan, V. Štrukil and T. Friščić, J. Am. Chem. Soc., 2015, 137, 2476 CrossRef CAS PubMed; (b) B. Rodríguez, A. Bruckmann and C. Bolm, Chem. – Eur. J., 2007, 13, 4710 CrossRef PubMed.
- (a) K. Užarević, I. Halasz, I. Đilović, N. Bregović, M. Rubčić, D. Matković-Čalogović and V. Tomišić, Angew. Chem., Int. Ed., 2013, 52, 5504 CrossRef PubMed; (b) T. Friščić, D. G. Reid, I. Halasz, R. S. Stein, R. E. Dinnebier and M. J. Duer, Angew. Chem., Int. Ed., 2010, 49, 712 CrossRef PubMed.
- W. C. Shearouse, M. Z. Shumba and J. Mack, Appl. Sci., 2014, 4, 171 CrossRef PubMed.
- T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66 CrossRef PubMed.
- (a) D. Gracin, V. Štrukil, T. Friščić, I. Halasz and K. Užarević, Angew. Chem., Int. Ed., 2014, 53, 6193 CrossRef CAS PubMed; (b) M. Juribašić, K. Užarević, D. Gracin and M. Ćurić, Chem. Commun., 2014, 50, 10287 RSC.
- L. Batzdorf, F. Fischer, M. Wilke, K.-J. Wenzel and F. Emmerling, Angew. Chem., Int. Ed., 2015, 54, 1799 CrossRef CAS PubMed.
- (a) G. Rothenerg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701 CrossRef PubMed; (b) O. Dolotko, J. W. Wiench, K. W. Dennis, V. K. Pecharsky and V. P. Balema, New J. Chem., 2010, 34, 25 RSC; (c) I. Halasz, A. Puškarić, S. A. J. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimäki, R. E. Dinnebier, B. Patel, W. Jones, V. Štrukil and T. Friščić, Angew. Chem., Int. Ed., 2013, 52, 11538 CrossRef CAS PubMed.
- S. L. Childs, N. Rodríguez-Hornedo, L. S. Reddy, A. Jayasankar, C. Maheshwari, L. McCausland, R. Shipplett and B. C. Stahly, CrystEngComm, 2008, 10, 856 RSC.
- K. S. McKissic, J. T. Caruso, R. G. Blair and J. Mack, Green Chem., 2014, 16, 1628 RSC.
- That the Curtius rearrangement is possible using this substrate was verified by heating the solution of acyl azide where isocyanate was formed.
- (a) V. Štrukil, B. Bartolec, T. Portada, I. Đilović, I. Halasz and D. Margetić, Chem. Commun., 2012, 48, 12100 RSC; (b) T.-X. Métro, J. Bonnamour, T. Reidon, J. Sarpoulet, J. Martinez and F. Lamaty, Chem. Commun., 2012, 48, 11781 RSC; (c) J. Bonnamour, T.-X. Métro, J. Martinez and F. Lamaty, Green Chem., 2013, 15, 1116 RSC; (d) I. Dokli and M. Gredičak, Eur. J. Org. Chem., 2015 DOI:10.1002/ejoc.20150005.
- I. Noda and Y. Ozaki, Two-Dimensional Correlation Spectroscopy - Applications in Vibrational and Optical Spectroscopy, John Wiley & Sons, 2004 Search PubMed.
- Full geometry optimization and vibrational calculations were performed using the B3LYP method as implemented in the Gaussian09 program.15b The standard 6-311G** basis set was used for C, H, N and O atoms; the calculated Raman spectra were scaled using scaling factors (0.975–0.983) which gave the best fit with the experimental data. M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- E. E. Ernstbrunner, R. B. Girling, W. E. L. Grossman, E. Mayer, K. P. J. Williams and R. E. Hester, J. Raman Spectrosc., 1981, 10, 161 CrossRef CAS PubMed.
- (a) E. A. Pritchina, N. P. Gritsan, A. Maltsev, T. Bally, T. Autrey, Y. Liu, Y. Wang and J. P. Toscano, Phys. Chem. Chem. Phys., 2003, 5, 1010 RSC; (b) J. Jiang, X. Wu, D. Li, B. Ma, R. Liu, X. Wang, J. Zhang, H. Zhu and Q. Cui, J. Phys. Chem. B, 2015, 119, 513 CrossRef CAS PubMed; (c) X. Zeng, E. Bernhardt, H. Beckers, K. Banert, M. Hagedorn and H. Liu, Angew. Chem., Int. Ed., 2013, 52, 3503 CrossRef CAS PubMed.
- (a) S. Shen and J. R. Durig, J. Mol. Struct., 2003, 661–662, 49 CrossRef CAS; (b) D. A. Dows and G. C. Pimentel, J. Chem. Phys., 1955, 23, 1258 CrossRef CAS PubMed; (c) J. Nelson, Spectrochim. Acta, Part A, 1970, 26, 235 CrossRef CAS.
- Mechanochemical reactions were carried out using a Retsch MM301 ball mill operating at 30 Hz and in-house manufactured 14 mL transparent poly(methylmetha)acrylate (PMMA) milling vessels with two 7 mm diameter stainless steel balls (ball weight 1.3 g). For each experiment, 266 mg (1.38 mmol) azide and 150 mg (1.38 mmol) amine were directly weighed to one half of the reaction vessel. The other half was filled with grinding balls and 75 μL of the liquid additive giving the same ratio of liquid volume to mass of solid reactants (η) in all experiments, η = 0.18 μL mg−1. The two halves of the jar were carefully joined together so that the solids did not mix with the liquid before milling was initiated. In situ monitoring was performed as described before using a portable Raman spectrometer13 with a PD-LD LS2 laser source (784 nm). Scattered radiation was analysed using an Oceanoptics Maya2000 Pro spectrometer. The Raman probe for the laser was positioned below the milling vessel for data collection. Each spectrum was typically collected for 10 seconds. The PMMA vessel contribution to the collected Raman spectra was subtracted. Products were characterized by 1H and 13C NMR and infrared spectroscopy (Fig. S1–S4 in the ESI†) as well as by PXRD (Fig. S16 in the ESI†).
- V. Gutmann, Coord. Chem. Rev., 1976, 18, 225 CrossRef CAS.
- A. Delori, T. Friščić and W. Jones, CrystEngComm, 2012, 14, 2350 RSC . Also, for a pseudo-fluid model of milling reactions see: X. Ma, W. Yuan, S. E. J. Bell and S. L. James, Chem. Commun., 2014, 50, 1585 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details. See DOI: 10.1039/c5cc01915j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |